Chapter: Basic & Clinical Pharmacology : Agents Used in Anemias; Hematopoietic Growth Factors
VITAMIN B12 - Agents Used In Anemias
VITAMIN B12
Vitamin B12 (cobalamin) serves as
a cofactor for several essential biochemical reactions in humans. Deficiency of
vitamin B12 leads to
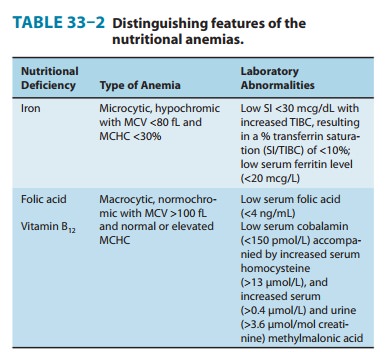
megaloblastic anemia (Table 33–2), gastrointestinal symptoms, and neurologic
abnormalities. Although deficiency of vitamin B12 due to an inadequate supply in the diet is
unusual, deficiency of B12 in adults—especially older adults—due to inadequate absorptionof
dietary vitamin B12 is a relatively common and easily treated disorder.
Chemistry
Vitamin B12 consists of a
porphyrin-like ring with a central cobalt atom attached to a nucleotide.
Various organic groups may be covalently bound to the cobalt atom, forming
different cobalamins. Deoxyadenosylcobalamin and methylcobalamin are the active
forms of the vitamin in humans. Cyanocobalamin
and hydroxo-cobalamin (both
available for therapeutic use) and other cobala-mins found in food sources are
converted to the active forms. The ultimate source of vitamin B12 is from microbial
synthesis; the vitamin is not synthesized by animals or plants. The chief
dietary source of vitamin B12 is microbially derived vitamin B12 in meat (especially liver), eggs, and dairy
products. Vitamin B12 is some-times called extrinsic
factor to differentiate it from intrinsicfactor,
a protein secreted by the stomach that is required forgastrointestinal
uptake of dietary vitamin B12.
Pharmacokinetics
The average American
diet contains 5–30 mcg of vitamin B12 daily, 1–5 mcg of which is usually absorbed.
The vitamin is avidly stored, primarily in the liver, with an average adult
having a total vitamin B12 storage pool of 3000–5000 mcg. Only trace amounts of vitamin B12 are normally lost in urine
and stool. Because the normal daily requirements of vitamin B12 are only about 2 mcg,
it would take about 5 years for all of the stored vitamin B12 to be exhausted and
for megaloblastic anemia to develop if B12 absorp-tion were stopped. Vitamin B12 is absorbed after it
complexes with intrinsic factor, a glycoprotein secreted by the parietal cells
of the gastric mucosa. Intrinsic factor combines with the vitamin B12 that is liberated
from dietary sources in the stomach and duode-num, and the intrinsic factor-vitamin
B12 complex is
subsequently absorbed in the distal ileum by a highly selective
receptor-mediated transport system. Vitamin B12 deficiency in humans most often results from
malabsorption of vitamin B12 due either to lack of intrinsic factor or to loss or
malfunction of the absorp-tive mechanism in the distal ileum. Nutritional
deficiency is rare but may be seen in strict vegetarians after many years
without meat, eggs, or dairy products.
Once absorbed, vitamin
B12 is transported to the
various cells of the body bound to a family of specialized glycoproteins,
transcoba-lamin I, II, and III. Excess vitamin B12 is stored in the liver.
Pharmacodynamics
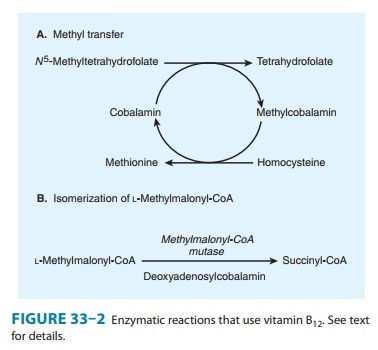
Two
essential enzymatic reactions in humans require vitamin B12
(Figure 33–2). In one, methylcobalamin serves as an intermediate in the
transfer of a methyl group from N5-methyltetrahydrofolate
to homocysteine, forming methionine (Figure 33–2A; Figure 33–3). Without
vitamin B12, conversion of the major dietary
and storage folate—N5-methyltetrahydrofolate—to
tetrahydrofolate, the precursor of folate cofactors, cannot occur. As a result,
vitamin B12 deficiency leads to deficiency of
folate cofactors necessary for several biochemical reactions involving the
transfer of one-carbon
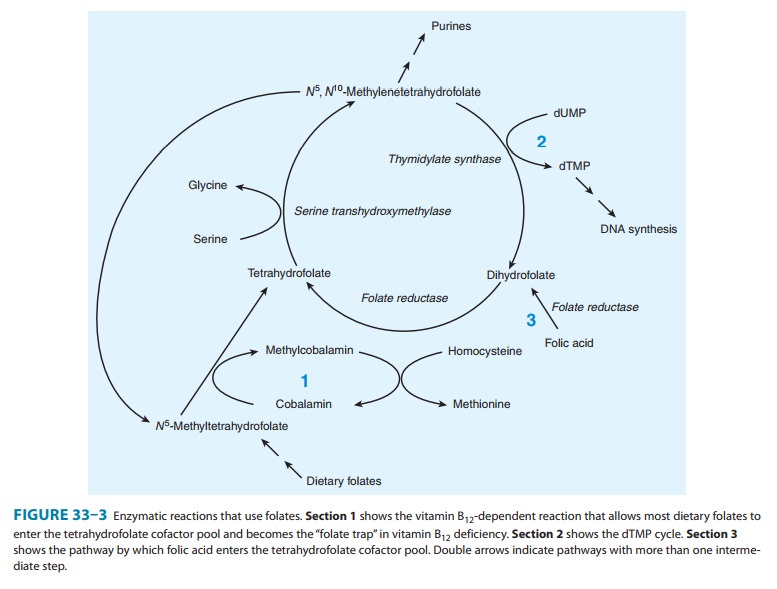
groups.
In particular, the depletion of tetrahydrofolate prevents synthesis of adequate
supplies of the deoxythymidylate (dTMP) and purines required for DNA synthesis
in rapidly dividing cells, as shown in Figure 33–3. The accumulation of folate
as N 5-methyltetrahydrofolate
and the associated depletion of tetra-hydrofolate cofactors in vitamin B12
deficiency have been referred to as the “methylfolate trap.” This is the
biochemical step whereby vitamin B12
and folic acid metabolism are linked, and it explains why the megaloblastic
anemia of vitamin B12 deficiency
can be partially corrected by ingestion of large amounts of folic acid. Folic
acid can be reduced to dihydrofolate by the enzyme dihydro-folate reductase
(Figure 33–3, section 3) and thereby serve as a source of the tetrahydrofolate
required for synthesis of the purines and dTMP required for DNA synthesis.
Vitamin
B12 deficiency causes the accumulation
of homo-cysteine due to reduced formation of methylcobalamin, which is required
for the conversion of homocysteine to methionine (Figure 33–3, section 1). The
increase in serum homocysteine can be used to help establish a diagnosis of
vitamin B12 deficiency (Table 33–2).
There is evidence from observational studies that elevated
serum homocysteine increases the risk of atherosclerotic cardiovascular
disease. However, randomized clinical trials have not shown a definitive
reduction in cardiovascular events (myocardial infarction, stroke) in patients
receiving vitamin supplementation that lowers serum homocysteine.
The
other reaction that requires vitamin B12
is isomerization of methylmalonyl-CoA to succinyl-CoA by the enzyme
methylmalonyl-CoA mutase (Figure 33–2B). In vitamin B12
deficiency, this con-version cannot take place and the substrate,
methylmalonyl-CoA, as well as methylmalonic acid accumulate. The increase in
serum and urine concentrations of methylmalonic acid can be used to support a
diagnosis of vitamin B12 deficiency
(Table 33–2). In the past, it was thought that abnormal accumulation of
methylmalonyl-CoA causes the neurologic manifestations of vitamin B12
defi-ciency. However, newer evidence instead implicates the disruption of the
methionine synthesis pathway as the cause of neurologic problems. Whatever the
biochemical explanation for neurologic damage, the important point is that
administration of folic acid in the setting of vitamin B12
deficiency will not prevent neurologic manifestations even though it will
largely correct the anemia caused by the vitamin B12
deficiency.
Clinical Pharmacology
Vitamin B12 is used to treat or
prevent deficiency. The most char-acteristic clinical manifestation of vitamin
B12 deficiency is
mega-loblastic, macrocytic anemia (Table 33–2), often with associated mild or
moderate leukopenia or thrombocytopenia (or both), and a characteristic
hypercellular bone marrow with an accumulation of megaloblastic erythroid and
other precursor cells. The neuro-logic syndrome associated with vitamin B12 deficiency usually
begins with paresthesias in peripheral nerves and weakness and progresses to
spasticity, ataxia, and other central nervous system dysfunctions. Correction
of vitamin B12 deficiency arrests
the progression of neurologic disease, but it may not fully reverse neurologic
symptoms that have been present for several months. Although most patients with
neurologic abnormalities caused by vitamin B12 deficiency have megaloblastic anemia when
first seen, occasional patients have few if any hematologic abnormalities.
Once a diagnosis of
megaloblastic anemia is made, it must be determined whether vitamin B12 or folic acid
deficiency is the cause. (Other causes of megaloblastic anemia are very rare.)
This can usually be accomplished by measuring serum levels of the vitamins. The
Schilling test, which measures absorption and uri-nary excretion of
radioactively labeled vitamin B12, can be used to further define the mechanism of vitamin B12 malabsorption when
this is found to be the cause of the megaloblastic anemia.
The
most common causes of vitamin B12
deficiency are perni-cious anemia, partial or total gastrectomy, and conditions
that affect the distal ileum, such as malabsorption syndromes, inflammatory
bowel disease, or small bowel resection.
Pernicious anemia results
from defective secretion of intrinsicfactor by the gastric mucosal cells.
Patients with pernicious anemia have gastric atrophy and fail to secrete
intrinsic factor (as well as hydrochloric acid). The Schilling test shows
diminished absorp-tion of radioactively labeled vitamin B12,
which is corrected when intrinsic factor is administered with radioactive B12,
since the vitamin can then be normally absorbed.
Vitamin
B12 deficiency also occurs when the
region of the dis-tal ileum that absorbs the vitamin B12-intrinsic
factor complex is damaged, as when the ileum is involved with inflammatory
bowel disease or when the ileum is surgically resected. In these situations,
radioactively labeled vitamin B12
is not absorbed in the Schilling test, even when intrinsic factor is added.
Rare cases of vitamin B12 deficiency
in children have been found to be secondary to con-genital deficiency of
intrinsic factor or to defects of the receptor sites for vitamin B12-intrinsic
factor complex located in the distal ileum.
Almost
all cases of vitamin B12 deficiency
are caused by malab-sorption of the vitamin; therefore, parenteral injections
of vitamin B12 are required for therapy. For
patients with potentially revers-ible diseases, the underlying disease should
be treated after initial treatment with parenteral vitamin B12.
Most patients, however, do not have curable deficiency syndromes and require
lifelong treat-ment with vitamin B12.
Vitamin
B12 for parenteral injection is
available as cyanocoba-lamin or hydroxocobalamin. Hydroxocobalamin is preferred
because it is more highly protein-bound and therefore remains longer in the
circulation. Initial therapy should consist of 100– 1000 mcg of vitamin B12
intramuscularly daily or every other day for 1–2 weeks to replenish body stores.
Maintenance therapy con-sists of 100–1000 mcg intramuscularly once a month for
life. If neurologic abnormalities are present, maintenance therapy injec-tions
should be given every 1–2 weeks for 6 months before switch-ing to monthly
injections. Oral vitamin B12-intrinsic
factor mixtures and liver extracts should not be used to treat vitamin B12
deficiency; however, oral doses of 1000 mcg of vitamin B12
daily are usually sufficient to treat patients with pernicious anemia who
refuse or cannot tolerate the injections. After pernicious anemia is in
remission following parenteral vitamin B12
therapy, the vitamin can be administered intranasally as a spray or gel.
Related Topics