Chapter: Basic & Clinical Pharmacology : Agents Used in Anemias; Hematopoietic Growth Factors
Folic Acid - Agents Used In Anemias
FOLIC ACID
Reduced forms of folic
acid are required for essential biochemical reactions that provide precursors
for the synthesis of amino acids, purines, and DNA. Folate deficiency is
relatively common, even though the deficiency is easily corrected by
administration of folic acid. The consequences of folate deficiency go beyond
the prob-lem of anemia because folate deficiency is implicated as a cause of
congenital malformations in newborns and may play a role in vascular disease
(see Box: Folic Acid Supplementation: A Public Health Dilemma).
Chemistry
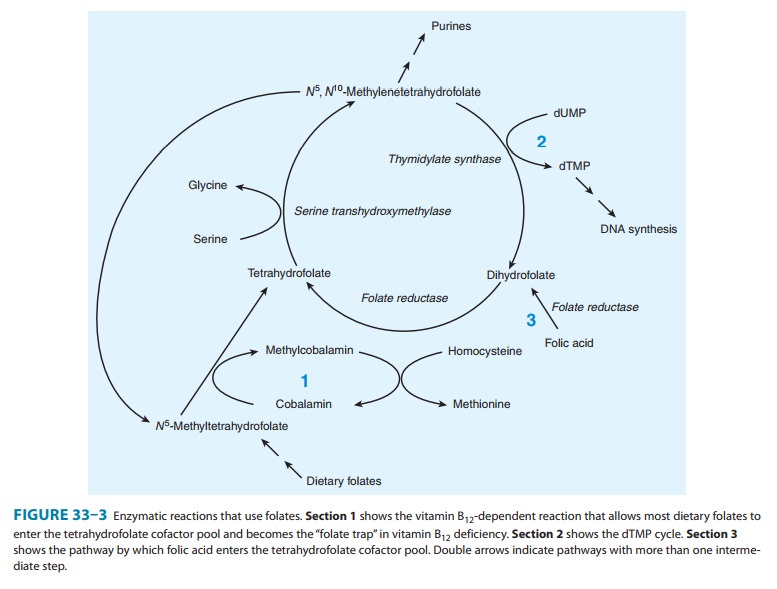
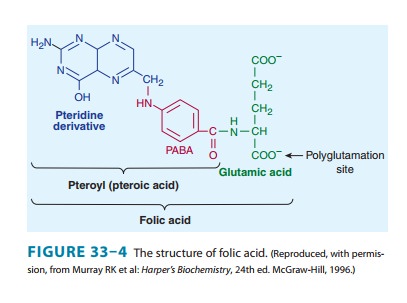
Folic acid (pteroylglutamic acid) is composed of a heterocycle (pteridine), p-aminobenzoic acid, and glutamic acid (Figure 33–4). Various numbers of glutamic acid moieties are attached to the pteroyl portion of the molecule, resulting in monoglutamates, triglutamates, or polyglutamates. Folic acid undergoes reduction, catalyzed by the enzyme dihydrofolate reductase (“folate reductase”), to give dihydrofolic acid (Figure 33–3). Tetrahydrofolate is subsequently transformed to folate cofactors possessing one-car-bon units attached to the 5-nitrogen, to the 10-nitrogen, or to both positions (Figure 33–3). Folate cofactors are interconvertible by various enzymatic reactions and serve the important biochemical function of donating one-carbon units at various levels of oxida-tion. In most of these, tetrahydrofolate is regenerated and becomes available for reutilization.
Pharmacokinetics
The average American
diet contains 500–700 mcg of folates daily, 50–200 mcg of which is usually
absorbed, depending on metabolic requirements. Pregnant women may absorb as
much as 300–400 mcg of folic acid daily. Various forms of folic acid are
present in a wide variety of plant and animal tissues; the richest sources are
yeast, liver, kidney, and green vegetables. Normally, 5–20 mg of folates is
stored in the liver and other tissues. Folates are excreted in the urine and
stool and are also destroyed by catabolism, so serum levels fall within a few
days when intake is diminished. Because body stores of folates are relatively
low and daily requirements high, folic acid deficiency and megaloblastic anemia
can develop within 1–6 months after the intake of folic acid stops, depending
on the patient’s nutritional status and the rate of folate utilization.
Unaltered folic acid
is readily and completely absorbed in the proximal jejunum. Dietary folates,
however, consist primarily of polyglutamate forms of N5-methyltetrahydrofolate.
Before absorp-tion, all but one of the glutamyl residues of the polyglutamates
must be hydrolyzed by the enzyme α-1-glutamyl transferase (“conjugase”) within
the brush border of the intestinal mucosa. The monoglutamate N5-methyltetrahydrofolate is subsequently
transported into the bloodstream by both active and passive transport and is
then widely distributed throughout the body. Inside cells, N5-methyltetrahydrofolate
is converted to tetrahydro-folate by the demethylation reaction that requires
vitamin B12 (Figure 33–3).
Pharmacodynamics
Tetrahydrofolate
cofactors participate in one-carbon transfer reac-tions. As described earlier
in the discussion of vitamin B12,
one of these essential reactions produces the dTMP needed for DNA synthe-sis.
In this reaction, the enzyme thymidylate synthase catalyzes the transfer of the
one-carbon unit of N5,
N10-methylenetetrahydrofolate
to deoxyuridine monophosphate (dUMP) to form dTMP (Figure 33–3, section 2).
Unlike all the other enzymatic reactions that use folate cofactors, in this
reaction the cofactor is oxidized to dihydrofolate, and for each mole of dTMP
produced, 1 mole of tetrahydrofolate is consumed. In rapidly proliferating
tissues, con-siderable amounts of tetrahydrofolate are consumed in this
reac-tion, and continued DNA synthesis requires continued regeneration of
tetrahydrofolate by reduction of dihydrofolate, catalyzed by the enzyme
dihydrofolate reductase. The tetrahydrofolate thus pro-duced can then reform
the cofactor N5,
N10-methylenetetrahydro-folate
by the action of serine transhydroxymethylase and thus allow for the continued
synthesis of dTMP. The combined catalytic activities of dTMP synthase,
dihydrofolate reductase, and serine transhydroxymethylase are referred to as
the dTMP synthesis cycle. Enzymes in
the dTMP cycle are the targets of two anticancer drugs; methotrexate inhibits
dihydrofolate reductase, and a metabolite of 5-fluorouracil inhibits
thymidylate synthase .
Cofactors
of tetrahydrofolate participate in several other essential reactions. N5-Methylenetetrahydrofolate
is required for the vitamin B 12-dependent
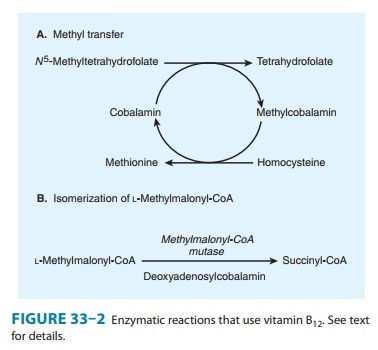
reaction that generates methionine from homocysteine (Figure 33–2A; Figure
33–3, section 1). In addition, tetrahydrofolate cofactors donate one-carbon
units dur-ing the de novo synthesis of essential purines. In these reactions,
tetrahydrofolate is regenerated and can reenter the tetrahydrofo-late cofactor
pool.
Clinical Pharmacology
Folate deficiency results
in a megaloblastic anemia that is micro-scopically indistinguishable from the
anemia caused by vitamin B12 deficiency (see above). However, folate
deficiency does not cause the characteristic neurologic syndrome seen in
vitamin B12 deficiency. In patients with megaloblastic anemia,
folate status is assessed with assays for serum folate or for red blood cell
folate. Red blood cell folate levels are often of greater diagnostic value than
serum levels, because serum folate levels tend to be labile and do not
necessarily reflect tissue levels.
Folic acid deficiency is
often caused by inadequate dietary intake of folates. Patients with alcohol
dependence and patients with liver disease can develop folic acid deficiency
because of poor diet and diminished hepatic storage of folates. Pregnant women
and patients with hemolytic anemia have increased folate requirements and may
become folic acid-deficient, especially if their diets are marginal. Evidence
implicates maternal folic acid deficiency in the occurrence of fetal neural
tube defects. (See Box: Folic Acid Supplementation: A Public Health Dilemma.)
Patients with malabsorption syn-dromes also frequently develop folic acid
deficiency. Patients who require renal dialysis are at risk of folic acid
deficiency because folates are removed from the plasma during the dialysis
procedure.
Folic acid deficiency can
be caused by drugs. Methotrexate and, to a lesser extent, trimethoprim and
pyrimethamine, inhibit dihydrofolate reductase and may result in a deficiency
of folate cofactors and ultimately in megaloblastic anemia. Long-term therapy
with phenytoin can also cause folate deficiency, but only rarely causes
megaloblastic anemia.
Parenteral administration
of folic acid is rarely necessary, since oral folic acid is well absorbed even
in patients with malabsorption syndromes. A dose of 1 mg folic acid orally
daily is sufficient to reverse megaloblastic anemia, restore normal serum
folate levels, and replenish body stores of folates in almost all patients.
Therapy should be continued until the underlying cause of the deficiency is
removed or corrected. Therapy may be required indefinitely for patients with
malabsorption or dietary inadequacy. Folic acid supplementation to prevent
folic acid deficiency should be consid-ered in high-risk patients, including
pregnant women, patients with alcohol dependence, hemolytic anemia, liver
disease, or cer-tain skin diseases, and patients on renal dialysis.
Related Topics