Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Mycobacteria
Tuberculosis : Pathogenesis
PATHOGENESIS
Primary Infection
Primary tuberculosis is the response to the initial infection in an individual not previously infected and sensitized to tuberculoprotein. Inhaled droplet nuclei containing small num-bers of tubercle bacilli are deposited in the peripheral respiratory alveoli, most frequently those of the well-ventilated middle and lower lobes. Here they are engulfed by nonspecif-ically activated alveolar macrophages. The ability of these cells to destroy ingested or-ganisms depends significantly on their inherent microbicidal capacity. If the alveolar macrophages are unable to destroy ingested mycobacteria, they continue to multiply until the macrophage bursts. The released organisms are subsequently ingested by inactivated blood macrophages that, together with T cells, are attracted to the lung by chemotactic factors.
The ingested mycobacteria continue to multiply intracellularly without damage to their host cell. Some of the bacterial-laden macrophages are transported through lym-phatic channels to the hilar lymph nodes draining the infected site. From there, they may disseminate through blood and lymphatic systems to a number of tissues, including the liver, spleen, kidney, bone, brain, meninges, and apices or other parts of the lung. The in-flammatory reaction in the seeded tissues is usually minor, and the signs and symptoms of infection are absent. However, the primary site of infection and some enlarged hilar lymph nodes can often be detected radiologically. In infants and immunocompromised adults, hematogenous dissemination of organisms may occasionally produce a life-threat-ening meningitis.
Morphologically, the resulting tubercle is a microscopic granuloma comprised of some multinucleated giant cells formed by the fusion of several macrophages (Langhans cells), many epithelioid cells (activated macrophages), and a surrounding collar of lym-phocytes (Fig 28 – 3) and fibroblasts. When many bacteria are present and there is a high degree of hypersensitivity, enzymes, reactive oxygen intermediates, and reactive nitrogen intermediates are released by dying macrophages and lead to necrosis of the center of the granuloma, which is termed caseous because of the cheesy, semisolid character of the gross lesion.
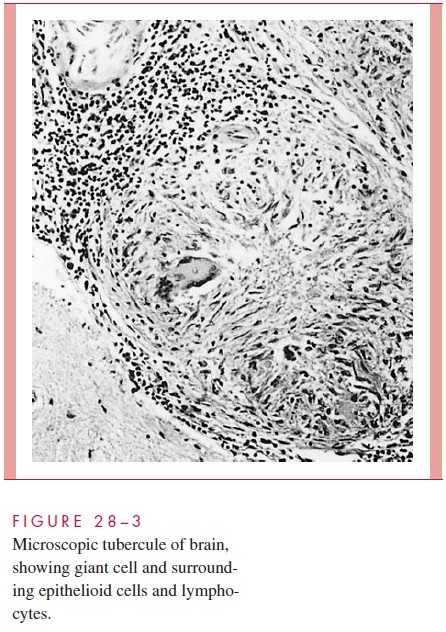
Primary infections are usually handled well by the host. Bacterial multiplication ceases. Most microscopic lesions heal by fibrosis, and the organisms in them slowly die. In others, especially those in well-oxygenated tissues such as the subapical areas of the lung, renal cortex and vertebral bodies, the tubercle bacilli remain viable for long periods and serve as a potential source of reactivation many months or years later if host defenses weaken.
Reactivation (Adult) Tuberculosis
Reactivation usually occurs in body areas of relatively high oxygen tension and low lym-phatic drainage, most often in the apex of the lung. The lesions show spreading, coalescing tubercles with numerous tubercle bacilli, and large areas of caseous necrosis. Necrosis often involves the wall of a small bronchus from which the necrotic material is discharged, result-ing in a pulmonary cavity and bronchial spread. Frequently, small blood vessels are also eroded. The chronic fever and weight loss may be mediated in part by macrophage-derived tumor necrosis factor.
Virulence Mechanisms
The basis for M. tuberculosis virulence is largely unknown. It produces no exotoxins, and both the intact cell and cellular components are remarkably innocuous to humans and ex-perimental animals not previously sensitized to tuberculin. Cell wall components such as LAM have been implicated in binding to alveolar macrophages, utilizing surface fi-bronectin, mannose, or complement receptors (CR1, CR3). Once inside, multiple factors contribute to survival and continued multiplication. A number of genes have been identi-fied that are linked to virulence by enhancing survival in the macrophage or by influenc-ing the physical and chemical conditions (low pH, high lactic acid, high CO2) present in developing lesions, but their function remains unknown. Mycolic acids, sulfolipids, LAM, and proteins have been shown to disrupt phagosome – lysosome interactions and interfere with oxidative killing. LAM has also been shown to modulate cytokine produc-tion and downregulate other aspects of T-cell function including antigen presentation.
Related Topics