Chapter: Basic & Clinical Pharmacology : Development & Regulation of Drugs
The Food & Drug Administration - Evaluation in Humans
The Food & Drug
Administration
The
FDA is the administrative body that oversees the drug evalu-ation process in
the USA and grants approval for marketing of new drug products. To receive FDA
approval for marketing, the originating institution or company (almost always
the latter) must submit evidence of safety and effectiveness. Outside the USA,
the regulatory and drug approval process is generally similar to that in the
USA.As its name suggests, the FDA is also responsible for certain aspects of
food safety, a role it shares with the US Department of Agriculture (USDA).
Shared responsibility results in complica-tions when questions arise regarding
the use of drugs, eg, antibiot-ics, in food animals. A different type of
problem arises when so-called food supplements are found to contain active
drugs, eg, sildenafil analogs in “energy food” supplements.The FDA’s authority
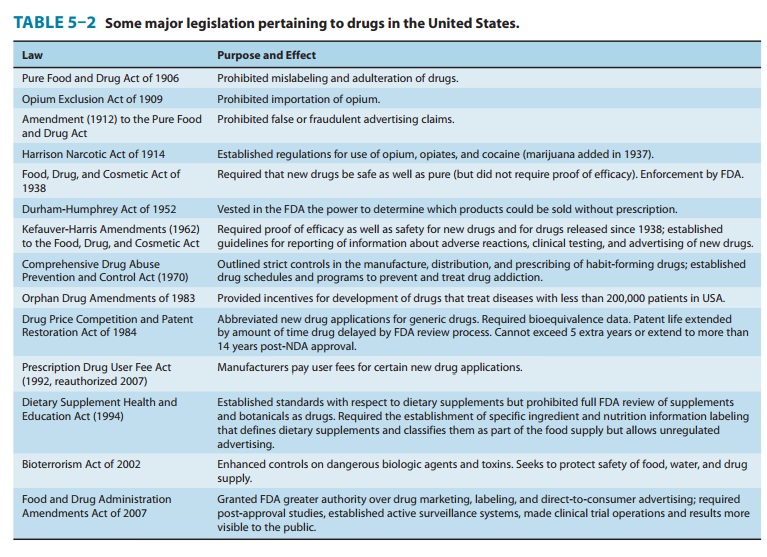
to regulate drugs derives from specific legislation (Table 5–2). If a drug has
not been shown through adequately controlled testing to be “safe and effective”
for a specific use, it cannot be marketed in interstate commerce for this use.∗Unfortunately, “safe” can mean different
things to the patient, the physician, and society. Complete absence of risk is
impossible to demonstrate, but this fact may not be understood by the public,
who frequently assume that any medication sold with the approval of the FDA
should be free of serious “side effects.” This confusion is a major factor in
litigation and dissatisfaction with aspects of drugs and medical care.The
history of drug regulation (Table 5–2) reflects several health events that
precipitated major shifts in public opinion. The Pure Food and Drug Act of 1906
became law in response to rev-elations of unsanitary and unethical practices in
the meat-packing industry. The Federal Food, Drug, and Cosmetic Act of 1938 was
largely a reaction to deaths associated with the use of a preparation of
sulfanilamide marketed before it and its vehicle were adequatelytested. The
Kefauver-Harris amendments of 1962 were, in part, the result of a teratogenic
drug disaster involving thalidomide. This agent was introduced in Europe in
1957–1958 and, based on animal tests then commonly used, was marketed as a
“nontoxic” hypnotic and promoted as being especially useful during preg-nancy.
In 1961, reports were published suggesting that thalido-mide was responsible
for a dramatic increase in the incidence of a rare birth defect called
phocomelia, a condition involving shorten-ing or complete absence of the arms
and legs. Epidemiologic stud-ies provided strong evidence for the association
of this defect with thalidomide use by women during the first trimester of
pregnancy, and the drug was withdrawn from sale worldwide. An estimated 10,000
children were born with birth defects because of maternal exposure to this one
agent. The tragedy led to the requirement for more extensive testing of new
drugs for teratogenic effects and stimulated passage of the Kefauver-Harris
Amendments of 1962, even though the drug was not then approved for use in the
USA. In spite of its disastrous fetal toxicity and effects in pregnancy,
thalidomide is a relatively safe drug for humans other than the fetus. Even the
most serious risk of toxicities may be avoided or managed if understood, and
despite its toxicity, thalidomide isnow approved by the FDA for limited use as
a potent immuno-regulatory agent and to treat certain forms of leprosy.
Related Topics