Chapter: Basic & Clinical Pharmacology : Development & Regulation of Drugs
Clinical Trials: The IND & NDA - Evaluation in Humans
Clinical Trials: The IND &
NDA
Once
a new drug is judged ready to be studied in humans, a Notice of Claimed
Investigational Exemption for a New Drug (IND) must be filed with the FDA
(Figure 5–1). The IND includes (1) information on the composition and source of
the drug, (2) chemical and manufacturing information, (3) all data from animal
studies, (4) proposed plans for clinical trials, (5) the names and credentials
of physicians who will conduct the clinical trials, and (6) a compilation of
the key data relevant to study of the drug in humans that has been made
available to investigators and their institutional review boards.
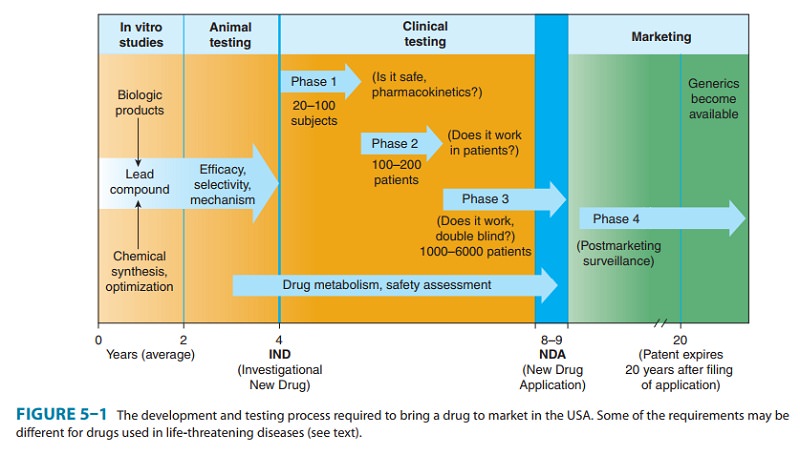
It
often requires 4–6 years of clinical testing to accumulate and analyze all
required data. Testing in humans is begun only after sufficient acute and
subacute animal toxicity studies have been completed. Chronic safety testing in
animals, including carcinoge-nicity studies, is usually done concurrently with
clinical trials. In each of the three formal phases of clinical trials,
volunteers or patients must be informed of the investigational status of the
drug as well as the possible risks and must be allowed to decline or to consent
to participate and receive the drug. These regulations are based on the ethical
principles set forth in the Declaration of Helsinki (1966). In addition to the
approval of the sponsoring organization and the FDA, an interdisciplinary
institutional review board (IRB) at the facility where the clinical drug trial
will be conducted must review and approve the scientific and ethical plans for
testing in humans.
In
phase 1, the effects of the drug as
a function of dosage are established in a small number (20–100) of healthy
volunteers. Although a goal is to find the maximum tolerated dose, the study is
designed to prevent severe toxicity. If the drug is expected to have
significant toxicity, as may be the case in cancer and AIDS therapy, volunteer
patients with the disease are used in phase 1 rather than normal volunteers.
Phase 1 trials are done to determine the prob-able limits of the safe clinical
dosage range. These trials may be nonblind or “open”; that is, both the
investigators and the subjects know what is being given. Alternatively, they
may be “blinded” and placebo controlled. The choice of design depends on the
drug, disease, goals of investigators, and ethical considerations. Many
predictable toxicities are detected in this phase. Pharmacokinetic measurements
of absorption, half-life, and metabolism are often done. Phase 1 studies are
usually performed in research centers by specially trained clinical
pharmacologists.
In
phase 2, the drug is studied in
patients with the target dis-ease to determine its efficacy (“proof of
concept”), and the doses to be used in any follow-on trials. A modest number of
patients (100–200) are studied in detail. A single-blind design may be used,
with an inert placebo medication and an established active drug (positive
control) in addition to the investigational agent. Phase 2 trials are usually
done in special clinical centers (eg, uni-versity hospitals). A broader range
of toxicities may be detected in this phase. Phase 2 trials have the highest
rate of drug failures, and only 25% of innovative drugs move on to phase 3.
In
phase 3, the drug is evaluated in
much larger numbers of patients with the target disease—usually thousands—to
further establish and confirm safety and efficacy. Using information gath-ered
in phases 1 and 2, phase 3 trials are designed to minimize errors caused by
placebo effects, variable course of the disease, etc. Therefore, double-blind
and crossover techniques are often used. Phase 3 trials are usually performed
in settings similar to those anticipated for the ultimate use of the drug.
Phase 3 studies can be difficult to design and execute and are usually
expensive because of the large numbers of patients involved and the masses of
data that must be collected and analyzed. The drug is formulated as intended
for the market. The investigators are usually specialists in the dis-ease being
treated. Certain toxic effects, especially those caused by immunologic
processes, may first become apparent in phase 3.
If
phase 3 results meet expectations, application is made for permission to market
the new agent. Marketing approval requires submission of a New Drug Application
(NDA)—or for biologicals, a Biological License Application—to the FDA. The
application contains, often in hundreds of volumes, full reports of all
preclini-cal and clinical data pertaining to the drug under review. The number
of subjects studied in support of the new drug application has been increasing
and currently averages more than 5000 patients for new drugs of novel structure
(new molecular entities). The duration of the FDA review leading to approval
(or denial) of the new drug application may vary from months to years. Priority
approvals are designated for products that represent significant improvements
compared with marketed products; in 2007, the median priority approval time was
6 months. Standard approvals, which take longer, are designated for products
judged similar to those on the market—in 2007, the median standard approval
time was 10.2 months. If problems arise, eg, unexpected but possibly serious
toxicities, additional studies may be required and the approval process may
extend to several years.
In
cases in which an urgent need is perceived (eg, cancer che-motherapy), the
process of preclinical and clinical testing and FDA review may be accelerated.
For serious diseases, the FDA may permit extensive but controlled marketing of
a new drug before phase 3 studies are completed; for life-threatening diseases,
it may permit controlled marketing even before phase 2 studies have been
completed. Roughly 50% of drugs in phase 3 trials involve early, controlled
marketing. Such “accelerated approval” is usually granted with the requirement
that careful monitoring of the effectiveness and toxicity of the drug be carried
out and reported to the FDA. Unfortunately, FDA enforcement of this requirement
has not always been adequate.
Once
approval to market a drug has been obtained, phase 4 begins. This constitutes monitoring the safety of the new
drug under actual conditions of use in large numbers of patients. The
importance of careful and complete reporting of toxicity by physi-cians after
marketing begins can be appreciated by noting that many important drug-induced
effects have an incidence of 1 in 10,000 or less and that some adverse effects
may become apparent only after chronic dosing. The sample size required to
disclose drug-induced events or toxicities is very large for such rare events.
For example, several hundred thousand patients may have to be exposed before
the first case is observed of a toxicity that occurs with an average incidence
of 1 in 10,000. Therefore, low-incidence drug effects are not generally
detected before phase 4 no matter how carefully the studies are executed. Phase
4 has no fixed dura-tion. As with monitoring of drugs granted accelerated
approval, phase 4 monitoring has often been lax.
The
time from the filing of a patent application to approval for marketing of a new
drug may be 5 years or considerably longer. Since the lifetime of a patent is
20 years in the USA, the owner of the patent (usually a pharmaceutical company)
has exclusive rights for marketing the product for only a limited time after
approval of the new drug application. Because the FDA review process can be
lengthy, the time consumed by the review is sometimes added to the patent life.
However, the extension (up to 5 years) cannot increase the total life of the
patent to more than 14 years after approval of a new drug application. As of
2005, the average effec-tive patent life for major pharmaceuticals was 11
years. After expiration of the patent, any company may produce the drug, file
an abbreviated new drug application (ANDA), demonstrate required equivalence,
and, with FDA approval, market the drug as a generic product without paying license fees to the original pat-ent
owner. Currently, 67% of prescriptions in the USA are for generic drugs. Even
biotechnology-based drugs such as antibodies and other proteins are now
qualifying for generic designation, and this has fueled regulatory concerns.
A
trademark is the drug’s proprietary
trade name and is usu-ally registered; this registered name may be legally
protected as long as it is used. A generically equivalent product, unless
specially licensed, cannot be sold under the trademark name and is often
designated by the official generic name.
The
FDA drug approval process is one of the rate-limiting fac-tors in the time it
takes for a drug to be marketed and to reach patients. The Prescription Drug
User Fee Act (PDUFA) of 1992, reauthorized in 2007, attempts to make more FDA
resources available to the drug approval process and increase efficiency
through use of fees collected from the drug companies that pro-duce certain
human drugs and biologic products. In 2009, the FDA approved 19 new molecular
entity drug applications for new nonbiologic entities and six biological
license applications, one more than in 2008. The traditional sequential and
linear drug development process previously described is being increasingly
modified in an attempt to safely accelerate clinical trials that pro-vide
“proof of mechanism” of action and “proof of concept” that the drug does work
in the target disease. In these newer approaches, certain development
activities such as full dose-response studies, final drug formulation work, and
long-term toxicology studies may be deferred. It is hoped that this approach
will focus resources on drugs more likely to succeed and minimize later-stage
failures. In one example, a phase 0 (phase zero) clinical trial is designed to
study the pharmacodynamic, pharmacokinetic properties of a drug and its links
to useful biomarkers and measures of mecha-nism. Unlike a phase 1 trial with
dose-response studies, in a phase 0 trial, a limited number of low doses are
administered. These tri-als are not designed to be therapeutic.
Related Topics