Chapter: Medical Immunology: Diagnostic Immunology
Tests Based on Immunofluorescence
Tests Based on Immunofluorescence
The primary reaction between antibodies chemically combined with fluorescent dyes and cell or tissue-fixed antigens can be visualized in a suitable microscope. Several variations of immunofluorescence can be used to detect the presence of unknown antigen in cells or tissues and the presence of unknown antibodies in patient’s serum (Fig. 15.10).
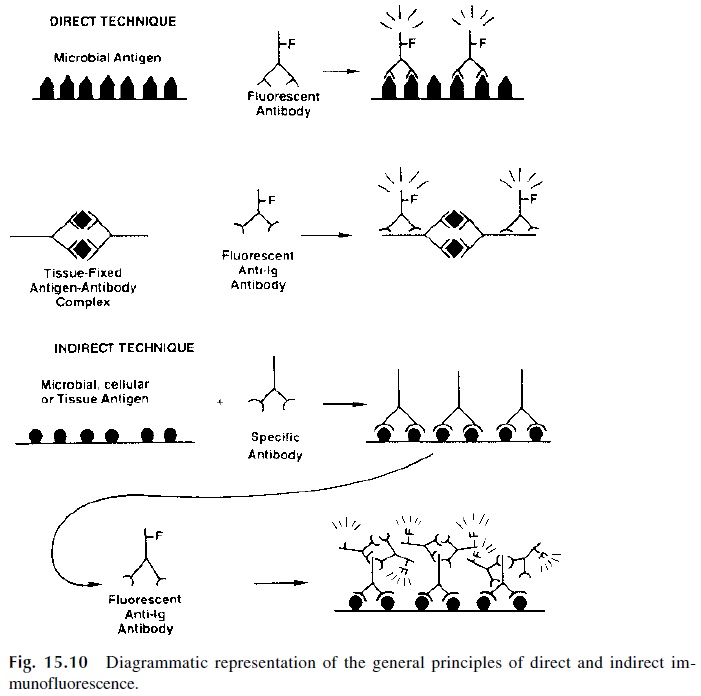
a. Direct immunofluorescence visualizes antigen in a cell or tissue involves direct labeling with fluorescent antibody. Similarly, tissue-deposited antigen-antibody complexes can be revealed by reaction with a fluorescent anti-immunoglobulin antibody.
b. Indirect immunofluorescence (as all indirect tests) involves two steps: (1) incu-bation of a substrate containing a fixed antigen (e.g., in a cell or tissue) with un-labeled antibody, which becomes associated with the antigen, and (2) after care-ful washing, a fluorescent antibody (e.g., fluorescent-labeled anti-IgG) is added to the substrate. This second antibody will become associated with the first, and the complex antigen-antibody 1-antibody 2 can be visualized on the fluorescence microscope. The indirect method has the advantage of using a single labeled an-tibody to detect many different specific antigen-antibody reactions.
1. Immunofluorescence Tests In Microbiology
Immunofluorescence has been widely applied in diagnostic tests. In microbiology it can be used to identify isolated organisms, to visualize infectious organisms in tissue biopsies or exudates, and to diagnose an infection through the demonstration of the corresponding an-tibodies.
A classical example is the use of the indirect fluorescence test for the diagnosis of syphilis. In a first step, the patient’s serum is incubated with killed Treponema pallidum; in the second step, a fluorescence-labeled anti-human antibody is used to determine if an-tibodies from the patient’s serum become bound to Treponema. Similar techniques have been used for the diagnosis of some viral diseases using virus-infected cells as a substrate.
Using fluorescent antibodies specific for different immunoglobulin isotypes, it is possible to identify the class of a given antibody after it has been captured on an antigen-coated solid phase. While IgG antibodies can be present in circulation for extended periods of time, IgM antibodies are characteristic of the early stages of the primary immune re-sponse. Thus, tests specifically designed to detect IgM antibodies are particularly useful for the diagnosis of ongoing infections.
2. Quantitative Immunofluorescence Assays
Quantitative immunofluorescence assays have been developed as a result of the introduc-tion of fluorometers. The principles are similar to those just mentioned: antigen is bound to a solid phase, exposed to a serum sample containing specific antibody, unbound im-munoglobulins are rinsed off, and a fluorescein-labeled antibody is added to reveal the an-tibody that reacted specifically with the immobilized antigen. A fluorometer is used to as-say the amount of fluorescence emitted by the second antibody. Since the amount of fluorescent antibody added to the system is fixed, the amount that remains bound is directly proportional to the concentration of antibody present in the sample. Thus, a quantitative correlation can be drawn between the intensity of fluorescence and the concentration of an-tibody added in the first step. These quantitative tests have been adapted to microbiologi-cal assays because they can combine quantitative and analytical properties (e.g., distin-guishing IgG from IgM antibodies).
3. Immunofluorescence Tests for the Detection of Autoantibodies
Immunofluorescence is also the technique of choice for the detection of autoantibodies such as antinuclear antibodies. Classically, the suspect serum is incubated with an adequate tissue (rat kidney, HeLa cells), and indirect immunofluorescence is performed in a second step to detect antibodies fixed to the substrate. In a positive test, the nuclei of the cells used as substrate will be fluorescent. The pattern of nuclear fluorescence is variable and has di-agnostic implications . Autoantibodies directed to a variety of tissue anti-gens can be detected by immunofluorescence, and this is the basis for a variety of tests used in the evaluation of a variety of organ-specific autoimmune diseases .
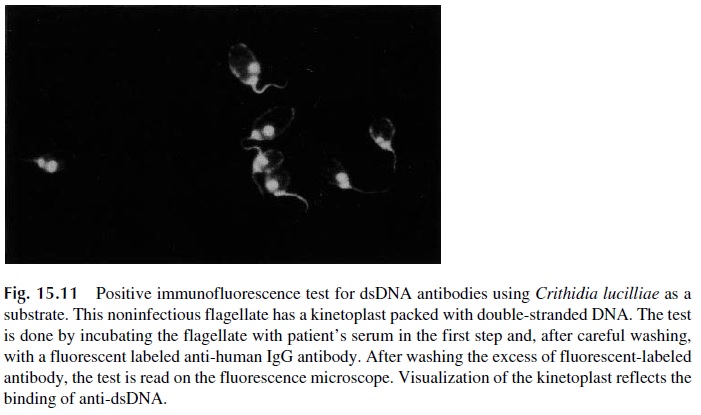
Anti-double-stranded (ds) DNA antibodies can also be detected by immunofluores-cence using a parasite (Chritidia lucilliae), which has a kinetoplast composed of pure ds DNA. Fixed parasites are incubated with patient’s serum in the first step. Anti-ds DNA an-tibodies, if present, will bind to the kinetoplast and will be revealed with fluorescein-la-beled anti-IgG antibody (Fig. 15.11).
4. Immunofluorescence Tests to Detect Tissue-Fixed Antigen- Antibody Complexes
Immunofluorescence assays have been extensively used by immunopathologists to detect immune complexes deposited in a variety of tissues. The technique usually involves performing a biopsy to obtain a tissue fragment that is then frozen and sectioned. The frozen sections are then incubated with fluorescence-labeled antibodies to immunoglobulins, C3, and fibrinogen (usually deposited in inflamed lesions).
5. Flow Cytometry
The development of murine hybridomas producing monoclonal antibodies to different cell markers has resulted in the development of reagents able to detect about 200 different cell membrane–associated antigenic systems. Many of those are expressed by lymphocytes, and a select few are known to identify subpopulations with different functions or on dif-ferent stages of activation . A variety of lymphocyte populations and sub-sets can be identified and enumerated by flow cytometry using fluorescent-labeled mono-clonal antibodies.
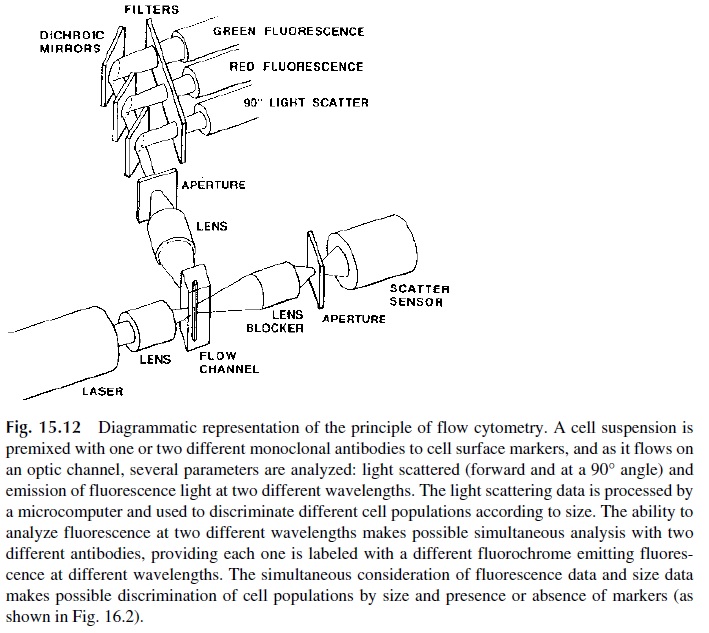
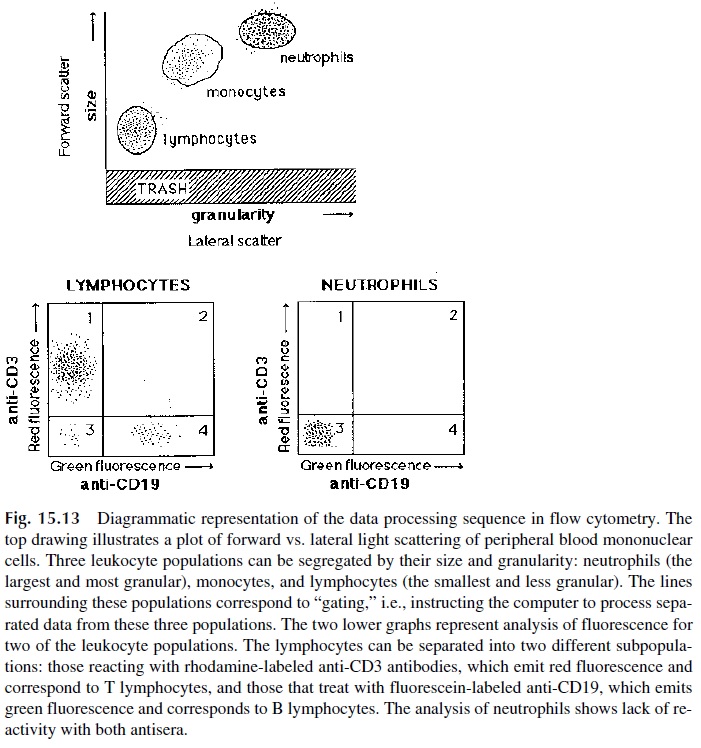
The principle of flow cytometry, as illustrated in Figure 15.12, is relatively simple. A diluted blood sample is incubated with one or several fluorescent-labeled monoclonal an-tibodies (if more than one antibody is used, each one is labeled with a different fluo-rochrome). The samples are further diluted so that they flow into a cell as a unicellular stream and are analyzed through light scattering and fluorescence. Light scattering mea-surements allow sizing of the cells (forward light scattering) and determination of their granularity (lateral light scattering, usually measured at 90°). Fluorescence measurements allow the determination of the number of cells expressing markers recognized by specific, fluorescent-labeled, monoclonal antibodies. By means of computer-assisted analysis, cell populations of homogeneous size can be segregated from cross-reactive cell types, which may express identical markers but be of a totally different nature, and analyzed for the ex-pression of one or several markers. Using monoclonal antibodies labeled with different flu-orochromes able to measure fluorescence at different wavelengths, it is possible to analyze cells for the simultaneous expression of three to five different markers (Fig. 15.13).
Surface Staining.The staining of membrane markers has found extensiveapplications in the study of quantitative and qualitative leukocyte abnormalities. The

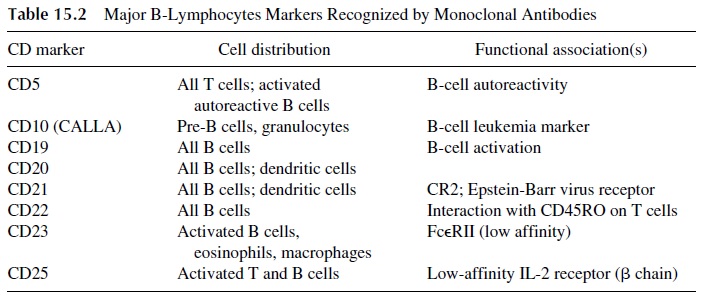

The study of T-lymphocyte subsets has found its main clinical application in the characterization of T-lymphocyte develop-ment abnormalities (e.g., primary immune deficiencies), acquired T-lymphocyte subpop-ulation deficiencies (e.g., AIDS), abnormalities in the quantitative distribution of T-lym-phocyte subpopulations (e.g., certain autoimmune diseases), and expression of markers not usually detected in normal peripheral blood lymphocytes (as in many types of leukemia).
CD19 and CD20 monoclonal antibodies are used to enumerate B cells. The fact that dendritic cells also express CD20 is inconsequential when phenotyping peripheral blood B lymphocytes. It should be noted that CD19, CD20, and MHC-II are not expressed by plasma cells; these cells express specific antigens, such as PCA-1.
Monocytes are usually identified by conventional staining on peripheral blood smears. When needed, these cells can be phenotyped on the basis of a combination of anti-gens, such as CD16 (common to all granulocytes and neutrophils), CD64 (high-affinity Fc receptor, unique to monocytes), and markers associated with the conserved regions of MHC-II (common to all antigen-presenting cells).
Phenotyping of NK cells with monoclonal antibodies is the most popular parameter to assess this cell subpopulation in a clinical context. However, the precise phenotype associated with a fully activated NK cell has not been established, and the enumeration of the resting NK cell population may not be very informative.
Cytoplasmic Staining.Flow cytometry can also be used to detect cytoplasmic pro-teins. A common example is the determination of cells containing terminal deoxynu-cleotidyl transferase (Tdt), a nuclear enzyme present in immature blood cells. It catalyzes the polymerization of deoxynucleotide triphosphates in the absence of a template. Its pres-ence, usually detected by immunofluorescence, is characteristic of immature T and B lym-phocytes and of the differentiated lymphocytes in leukemias and lymphomas. Its impor-tance is therefore related to the investigation of lymphocyte ontogeny and to the classification of lymphocytic malignancies.
Flow cytometry is also now commonly used for the detection of intracellular cy-tokine production. In addition to the cytokine measurement, with flow cytometric analysis the phenotype of the cytokine-producing cells can be evaluated.
DNA Analysis.Flow cytometric analysis of DNA index and cell cycle distributionhas clinical utility in the diagnosis and evaluation of cancers. The wide array of DNA-bind-ing fluorescent dyes, such as acridine orange (AO), propidium iodide (PI), Hoechst 33342, and 7-amino-actinomycin D (7-AAD), makes flow cytometry a powerful tool for nucleic acid analysis. Furthermore, flow cytometry is one of the preferred methods in the analysis of apoptosis. Flow cytometry is less labor intensive than other procedures for DNA analy-sis, and it allows for the simultaneous detection of cell surface phenotype and DNA content.
Sorting.Some flow cytometers have the ability to collect populations of cells (sort)or individual cells (clone) expressing a given phenotype, based on the expression of surface antigens. This powerful combination of analysis and isolation has been invaluable to the field of immunology research. In particular, the study of hematopoiesis and lymphopoiesis has benefited from this technology.
Researchers have been able to assess the differentiation potential of individual cloned progenitor cells in response to a variety of stimuli. Because the progenitor cells were sorted at the single-cell level (cloned), questions regarding con-taminating cell populations could not be raised. This approach has limitations, including time consumption, cost, sharing of markers by different cell populations, and relatively low yields.
A second approach that has become progressively more popular is magnetic sorting. This technique utilizes “magnetic” monoclonal antibodies, i.e., monoclonal antibodies to which magnetic microparticles have been chemically attached. After batch incubation of mononuclear cells with the magnetic monoclonal antibodies, the tube containing the mix-ture is placed in a magnetic field that attracts those cells with the marker recognized by the monoclonal antibody to the wall of the tube. Cells not recognized by the monoclonal anti-body remain in suspension and can be aspirated and further fractionated or discarded. To separate the cells retained on the tubes, the tubes are carefully rinsed while under the in-fluence of the magnetic field and then moved away from the magnetic field. The antibody-coated cells precipitate to the bottom of the tube, and the precipitated cells are then incu-bated at 37°C to dissociate them from the magnetized monoclonal antibody.
Related Topics