Chapter: Medical Immunology: Diagnostic Immunology
Precipitation Assays - Immunoserology
Precipitation Assays
Several of the commonly used serological assays are based on the detection of antigen-an-tibody aggregates, either through visualization of precipitates or by measuring the light dis-persed by antigen-antibody complexes in suspension.
1. Double Diffusion Method (Ouchterlony Technique)
This venerable and simple technique requires very little in terms of equipment and supplies other than antigens, antibodies, and a properly buffered agar gel. Wells are punched in an agar gel; antigen and antibody are placed in separate wells diffuse toward each other and precipitate at the point of antigen-antibody equivalence.
The main advantages of this technique are simplicity, minimal equipment require-ments, and specificity. If properly carried out, double immunodiffusion assays are 100% specific, as far as detecting an antigen-antibody reaction. The main limitations are lack of sensitivity and the time required for full development of visible precipitation (up to 72–96 hours of diffusion in systems when either reactant is present in small quantities).
In general, double diffusion assays are useful for determining the presence or absence of a given antigen or antibody in any kind of biological fluid. Some serological assays used in parasitology and mycology are based on double immunodiffusion.
2. Counterimmunoelectrophoresis
This technique is a variation of double immunodiffusion, in which antigen and antibody are forced to move towards each other with an electric current (antibodies move to the cathode, while most antigens have a strong negative charge and move towards the anode). Precip-itin lines can be visualized between the antigen and antibody wells.
This method is faster and more sensitive than double immunodiffusion. Visible pre-cipitation can be usually observed after 1–2 hours, although for maximal sensitivity it is necessary to wash, dry, and stain the agar gel in which the reaction took place, a process that extends the time to final reading of the results to several days.
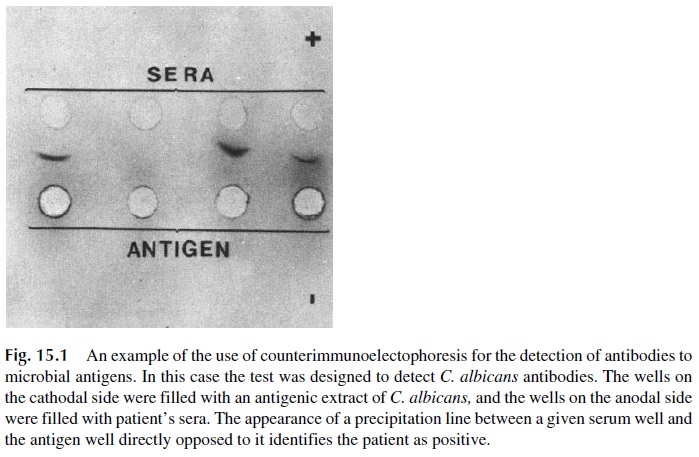
Counterimmunoelectrophoresis has been used for detection of fungal or bacterial antigens in cerebrospinal fluid (in patients with suspected meningitis) and for the detection of antibodies to Candida albicans(Fig. 15.1), DNA, and other antigens.
3. Radial Immunodiffusion
Radial immunodiffusion represents a hallmark in the evolution of immunoserology be-cause it represents the first successful attempt to develop a precise quantitative assay suit-able for routine use in the diagnostic laboratories. Radial immunodiffusion received its des-ignation from the fact that a given antigen is forced to diffuse concentrically on a support medium in which antiserum has been incorporated (Fig. 15.2).
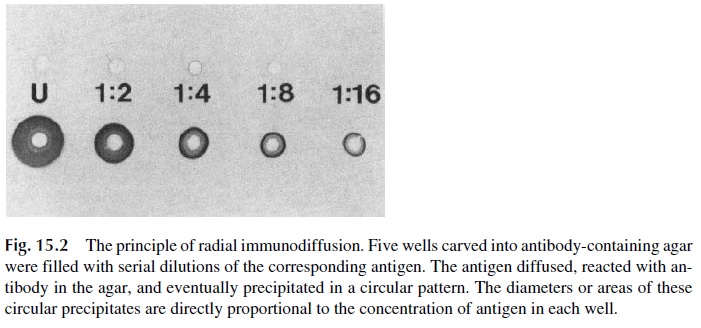
A polyclonal antiserum known to precipitate the antigen is added to molten agar, and an agar plate containing the antibody is then prepared. Identical wells are cut in the anti serum-containing agar, and those wells are filled with identical volumes of samples con-taining known amounts of the antigen (calibrators) and unknown samples in which the antigen needs to be assayed. After 24– 48 hours it is possible to measure the diameters of circular precipitates formed around the wells where antigens were placed. Those diameters are directly proportional to antigen concentration. A plot of precipitation ring diameters versus concentrations is made for the samples with known antigen concentrations. This plot, known as a calibration curve, is used to extrapolate the concentrations of antigen in the unknown samples based on the diameter of the corresponding precipitin rings.
Radial immunodiffusion, while not as sensitive as some newer techniques and rela-tively slow, is nonetheless reliable for routine assay of many serum proteins. It has been widely applied to the assay of immunoglobulins, complement components, and, in general, any antigenic protein that exists in concentrations greater than 0.5 mg/L in any biological fluid.
4. Quantitative Immunoelectrophoresis
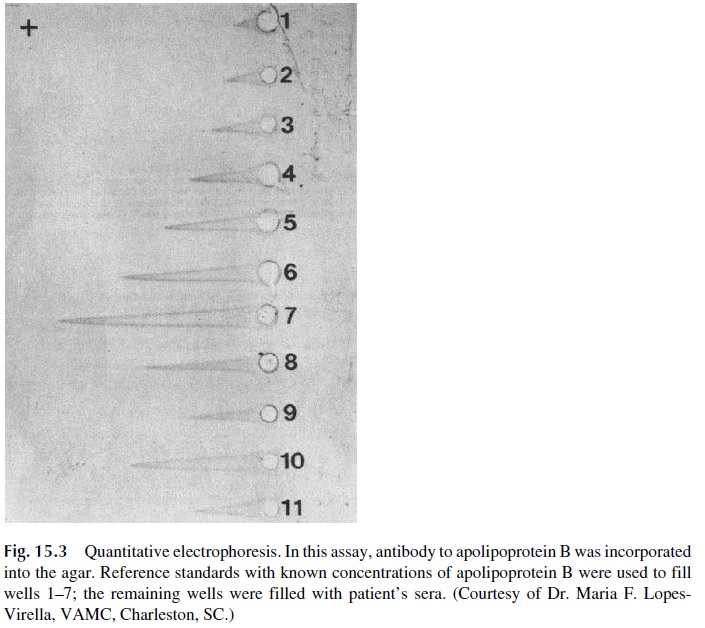
Quantitative immunoelectrophoresis (rocket electrophoresis) is an adaptation of radial im-munodiffusion in which the antigen is actively driven through an antibody-containing ma-trix by an electric potential. As the equivalence zone of antigen-antibody reaction is reached, the reaction ceases and elongated precipitin arcs (rockets) become visible (Fig. 15.3). The lengths of those rockets are proportional to the antigen concentrations in the wells. Rocket electrophoresis can be used for the quantitative assay of many proteins, in-cluding immunoglobulins. The reverse modality with antigen incorporated in the agar can be used for the measurement of specific antibodies. This technique is faster and more sen-sitive than radial immunodiffusion.
5. Immunonephelometry
This quantitative technique has replaced radial immunodiffusion, particularly when large numbers of samples are to be assayed. The principle of the assay lies in the fact that when dilute solutions of antigen and antibody are mixed under antibody excess conditions, soluble antigen-antibody complexes are formed that remain in suspension.
The relative amount of soluble aggregates can be assayed by measuring nephelometrically the amount of light dispersed by the mixture. This is most frequently done by passing a beam of light through tubes containing mixtures of fixed amounts of antibody and variable concentrations of anti-gen. The scatted light is measured at angles varying from near 0° to 45° relative to the in-cident light. If the antibody concentration is kept constant, the amount of dispersed light will be directly proportional to the antigen concentration. The principles of calibration and extrapolation of unknown values are identical to those of radial immunodiffusion.
Immunonephelometry is superior to radial immunodiffusion in terms of sensitivity, speed, and automation. However, the preparation of samples and cost of equipment and reagents make it mostly suitable for large-volume assays. In the right set-up, it is the tech-nique of choice for the measurement of immunogenic proteins (e.g., immunoglobulins or complement components) in concentrations greater than 5–10 mg/L when the volume of samples to be analyzed is large.
6. Immunoelectrophoresis
Immunoelectrophoresis (IEP) is a two-step technique that combines electrophoretic sepa-ration and double immunodiffusion. In the first step a protein mixture (such as serum) is electrophoresed through a support medium, usually agarose. After electrophoresis is com-pleted, specific antisera are deposited in a trough cut into the agarose, parallel to the axis of the electrophoretic separation. The electrophoretically separated proteins and the antisera diffuse toward one another, and at the zone of antigen-antibody equivalence, precipitin pat-terns in the form of arcs appear. This technique was widely used for analytical studies in patients suspected of plasma cell malignancies. It has been largely replaced by im-munoblotting methods, the interpretation of which is much simpler.
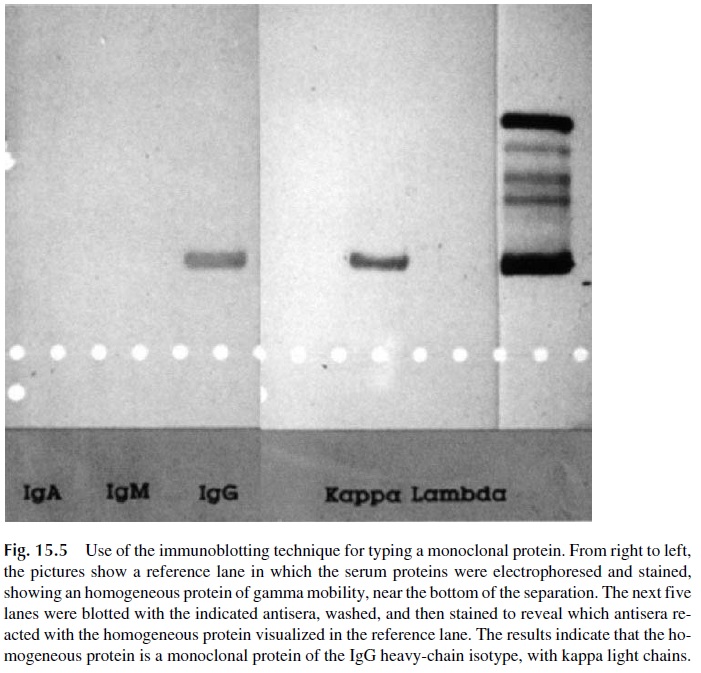
7. Immunofixation (Immunoblotting)
The principle of immunofixation is diagrammatically represented in Figure 15.4. In the first step, several aliquots of the patient’s serum are simultaneously separated by electrophore-sis. One of the separation lanes is stained as a reference for the position of the different serum proteins, while paper strips embedded with different antibodies are laid over the re-maining separation lanes. The antibodies diffuse into the agar and react with the corre-sponding immunoglobulins. After washing off unbound immunoglobulins and antibodies, the lanes where immunofixation takes place are stained, revealing whether the antisera rec-ognized the proteins they are directed against.
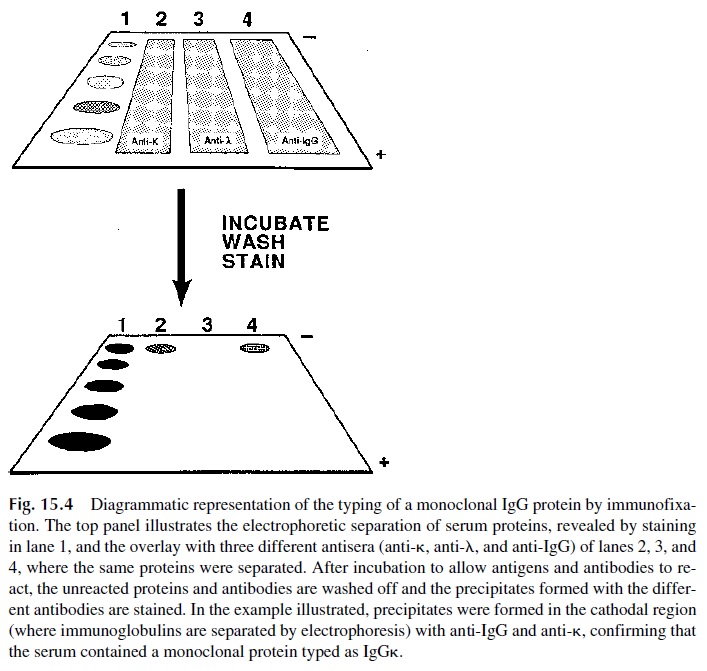
The main application of immunoblotting is the detection of monoclonal proteins. In the case illustrated in Figure 15.5, a patient’s serum was being tested for the presence of an IgG monoclonal protein. Such protein, by definition, must be homogeneous in mobility and must react with anti-IgG antibodies and either antikappa or antilambda antibodies. The di-agram shows stained precipitates corresponding to the lanes overlaid with anti-IgG and with anti–k light chains. This result proved that an IgG monoclonal protein existed in this patient’s serum.
8. Western Blot
The Western blot (Fig. 15.6) is a variation of immunoblotting that has been popularized by its use in the diagnosis of HIV infection. The first step in the preparation of an immunoblot

This is achieved by performing electrophoresis in the presence of a nega-tively charged detergent (such as sodium dodecyl sulfate), which becomes associated with the proteins and obliterates their charge differences, using as support for the separation a medium with sieving properties. The result is the separation of a protein mixture into com-ponents of different sizes.
After the separation of the viral proteins is completed, it is necessary to transfer the separated proteins to another support in order to proceed with the remaining steps. This transfer or “blotting” is easily achieved by forcing the proteins to migrate into a nitrocellu-lose membrane by a second electrophoresis step (electroblotting). The nitrocellulose mem-brane, to which the viral antigens have been transferred, is then impregnated with patient’s serum. If antibodies to any or several of these antigens are contained in the serum, they will combine with the antigen at the point where it is blotted.
The following steps are designed to detect the antigen-antibody complexes formed in the cellulose membrane. First, all the nonprecipitated antigens and antibodies are washed off; then the protein binding sites still available on the membrane are blocked to prevent false-positive reactions; finally, the membrane is overlaid with a labeled antibody to human immunoglobulins.
This antibody will react with the complexes formed by viral antigens and human antibodies and can later be revealed either by adding a color- or chemilumines-cence-developing substrate (if the anti-human immunoglobulin is labeled with an enzyme) or by autoradiography (if the anti-human immunoglobulin is labeled with 125iodine). The retention of the labeled antibody in site(s) known to correspond to the separation of viral proteins indicates that the patient’s serum contained antibodies reacting with those anti-gens.
This technique has the advantage over other screening assays of not only detecting antibodies, but identifying the antigens against which the antibodies are directed. This re-sults in increased specificity. The downside is the time consumed running the assay, which requires very careful quality control and needs to be carried out by specialized personnel in certified laboratories. To circumvent this problem, prepared Western blot membranes can be purchased commercially. These specific blots (e.g., for HIV) need only be probed with the patient serum and developed. Generally, positive and negative controls are included with the blots. Using these commercially available kits, the performance of sophisticated technology is accomplished with less time and training.
Related Topics