Agarose Gel Electrophoresis, Polymerase Chain Reaction (PCR) - Techniques in Genetic Engineering | 12th Microbiology : Chapter 12 : Microbial Genetics
Chapter: 12th Microbiology : Chapter 12 : Microbial Genetics
Techniques in Genetic Engineering
Techniques in Genetic Engineering
There are
several techniques used in recombinant DNA technology or gene manipulation. The
most frequently used methods are agarose gel electrophoresis, isolation and
purification of nucleic acids, nucleic acid blotting techniques, DNA
sequencing, chemical synthesis of DNA, gene transfer methods, polymerase chain
reaction, construction of gene library, radiolabeling of nucleic acids etc, few
of them are discussed here.
Agarose Gel Electrophoresis
Electrophoresis
refers to the movement of charged molecules in an electric field. The
negatively charged molecules move towards the positive electrode while the
positively charged molecules migrate towards the negative electrode. Gel
electrophoresis is a routinely used analytical technique for the separation and
purification of specific DNA fragments. The gel is composed of either
polyacrylamide or agarose. Polyacrylamide gel electrophoresis (PAGE) is used
for the separation of smaller DNA fragments while agarose electrophoresis is
convenient for the separation of DNA fragments ranging in size from 100 base
pairs to 20 kilobase pairs. Gel electrophoresis can also be used for the
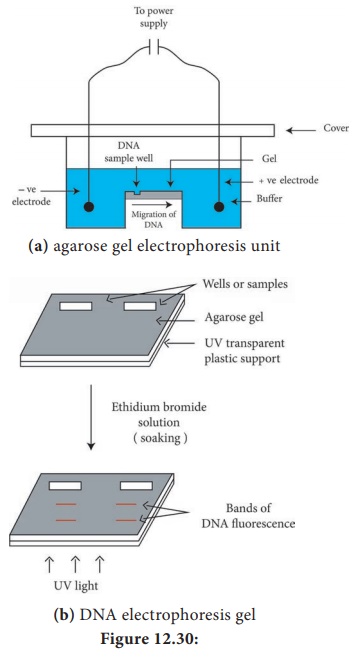
separation of RNA molecules. A diagramatic view of the agarose gel
electrophoresis unit is shown in Figure 12.30a
A
genomic library is a collection of clones that contains
at least one copy of every DNA sequence in an organism’s genome. Like libraries
with books, genomic libraries are a great source of information; in this case,
the information is about the genome. Specific sequences in cDNA libraries and
genomic libraries can be identified via a number of approaches, including the
use of specific antibodies, cDNA probes and oligonucleotide probes
Human artificial chromosome (HAC)-based vectors offer a
promising system for delivery and expression of full-length human genes of any
size into human cells, and a tool for determining human chromosome function. It
does not have the problem of limited cloning capacity of other vectors, and it
also avoids possible insertional mutagenesis caused by integration into host
chromosomes by viral vector.
Steps
1. Gel is
set with wells on one end.
2. The
gel is placed in an electrophoresis apparatus and covered with buffer solution.
3. The
DNA samples along with tracer dye are placed in the wells of gel
4. Power
supply is switched on and gel is run till the tracer dye reaches the end of the
gel.
As the DNA is negatively charged, DNA fragments
move through the gel towards the positive electrode. The rate of migration of
DNA is dependent on the size and shape. In general, smaller linear fragments
move faster than the larger ones. Hence, gel electrophoresis can be
conveniently used for the separation of a mixture of DNA fragments, based on
their size. The bands of the DNA can be detected by soaking the gel in ethidium
bromide solution (Ethidium bromide can also be added to molten agarose prior to
setting the gel). When activated by ultraviolet radiation, DNA base pairs in
association with ethidium bromide, emit orange fluorescence. And in this way
the DNA fragments separated in agarose electrophoresis can be identified
(Figure 12.30b).
PAGE is composed of chains of acryl amide monomers crosslinked with methylene bisacryalmide units. The pore size of the gel is dependent on the total concentration of monomers and the cross links. PAGE is used for the separation of single stranded DNA molecules that differ in length by just one nucleotide. Agarose gels cannot be used for this purpose. This is because polyacrylamide gels have smaller pore sizes than agarose gels and allow precise separation of DNA molecule from 10–1500 bp.
HOTS: Explain how gel electrophoresis can
be used to determine the size of a PCR product.
Polymerase Chain Reaction (PCR)
The PCR
technique has already proven exceptionally valuable in many areas of molecular
biology, medicine, and biotechnology. PCR technique has great practical
importance and impact on biotechnology. Between 1983 and 1985 American
biochemist Kary Mullis developed PCR technique that made it possible to
synthesize large quantities of a DNA fragment without cloning it. Mullis
received the 1993 Nobel Prize for Chemistry for his invention. PCR is a cell
free amplification technique.
Figure 12.31 outlines how PCR technique works. To amplify (make large quantities) a particular DNA sequence by PCR a reaction mixture (often 100μl or less in volume) containing the following are required.
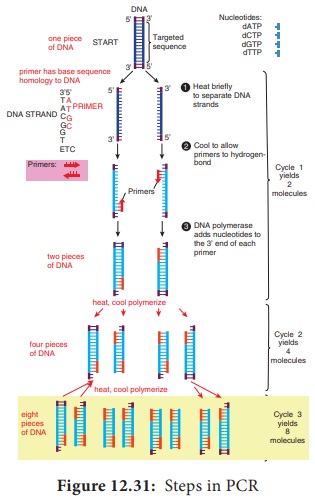
1. Target
DNA
2. Two primers–These are synthetic
oligonucleotides, usually about 20 nucleotides long. These are fragments
with sequences identical to those flanking the targeted sequence.
3. Thermostable DNA polymerase– Two popular enzymes employed in the PCR
technique are Taq polymerase from the thermophilic bacterium Thermus aquaticus and the vent polymerase from Theromococcus litoralis.
These polymerases employed in PCR technique are able to function at high
temperatures.
4. Four
deoxyribonucleoside triphosphates (dNTPs)- dCTP, dATP, dGTP, dTTP
Infobits
Cloned genes and other DNA sequences
often are analyzed to determine the arrangement and specific locations of
restriction sites. The analytical process involves cleavage of the DNA with
restriction enzymes, followed by separation of the resulting DNA fragments by
agarose gel electrophoresis. The sizes of the DNA fragments are calculated,
enabling restriction maps to be constructed. The many DNA fragments produced by
cleaving genomic DNA show a wide range of sizes, resulting in a continuous
smear of DNA fragments in the gel. In this case, specific gene fragments can be
visualized only by transferring them to membrane filter by southern blotting,
hybridizing a specific labelled probe with the DNA fragments, and detecting the
hybrids. A similar procedure, Northern blotting is used to analyze the sizes
and quantities of RNAs isolated from cell.
Taq polymerase proof reading exonuclease (3′–5′)
activity which might contribute to errors in the products of PCR. Some other
thermostable DNA polymerases with proof reading activity have been identified.
Example: Tma DNA polymerase from Thermotoga maritana.
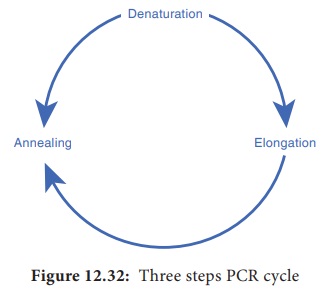
Steps in PCR
1. Denaturation:
The target
DNA containing the sequence to be
amplified is heat denatured to separate its complementary strands at
temperature 94 °C–95 °C.
2. Annealing:
The temperature is lowered to 37
°C–55 °C so that the primers can hydrogen bond or anneal to the DNA on both sides
of the target sequence. Because the primes are present in excess the targeted
DNA strands normally anneal to the primers rather than to each other.
3. Extension: Heat resistant DNA polymerase extends the primers and
synthesizes copies of the target DNA sequence using the deoxyribonucleoside
triphosphate’s at 70 °C–75 °C
The three
– step cycle (Figure 12.32) is repeated to obtain copies of target DNA in large
numbers. At the end of one cylcle, the targeted sequences on both strands have
been copied. When the three step cycle is repeated, the four strands from the
first cycle are copied to produce eight fragments. The third cycle yields 16
products. Theoretically, 20 cycles will. produce about one million copies of
the target DNA sequence. Each cycle of PCR takes about 3 – 5 minutes.
The PCR
technique has now been automated and is carried out by a specially designed
machine (Figure 12.33) PCR machines are now fully automated and microprocessor
controlled. They can process up to 96 samples at a time. PCR machines can carry
out 25 cycles and amplify DNA 105 times in as little as 57 minutes.

The PCR has many applications in research and
in commercial arena, including generating specific DNA segments for cloning or
sequencing, amplifying DNA to detect specific genetic defects, and amplifying
DNA for fingerprinting in crime scene investigation.
PCR technology is improving continually. Various forms of PCR are available. RNA too can be efficiently used in PCR procedures. Cellular RNAs and RNA viruses may be studied even when the RNA is present in very small amounts (as few as 100 copies can be transcribed and amplified). Quantitative PCR is quite valuable in virology and gene impression studies. PCR is modified as per the specific demands of the situation. Thus there are many variations in the original PCR Examples nested PCR, inverse PCR, reverse transcription PCR, time quantitative PCR, RAPD, RFLP, AFLP.
HOTS
Both PCR and Cloning allow for the production of many copies of
a DNA sequence. What are the advantages of using PCR instead of cloning to
amplify a DNA template?
What advantages are there to using a
DNA polymerase for PCR that has proofreading activity?
Infobits
In 1970s American molecular
biologists Allan M. Maxam and Walter Gilbert and English biochemist Frederick
Sanger developed some of the first techniques for DNA sequencing. Gilbert and
Sanger shared the 1980 Nobel Prize for Chemistry for their work. Dideoxy
procedure is one of the procedure used to sequence DNA.
Related Topics