Chapter: Biotechnology: Genomics and Bioinformatics
Sequences and nomenclature - Bioinformatics
Sequences and nomenclature
The nomenclature system we adopt in Bioinformatics work is based on the International Union of Pure and Applied Chemistry (IUPAC) recommendations. It is useful to follow this nomenclature system so that data sets from different laboratories situated around the world can be compared easily and uniformly. The database institutions and the journals that publish research reports follow these recommendations strictly to ensure uniformity and to aid rapid reproducibility. We will go through the basic nomenclature system for nucleic acids and proteins in this section. Details of modifications of the nucleotides may be touched upon but we suggest you refer to the IUPAC website for these details. For routine work using the nucleic acid and protein sequence data we discuss the following system of IUPAC nomenclature (Fig. 7).

Fig. 7. Comparison between Human communication and biological system.
DNA and protein sequences
Table 3. Summary of single-letter code IUPAC recommendations.
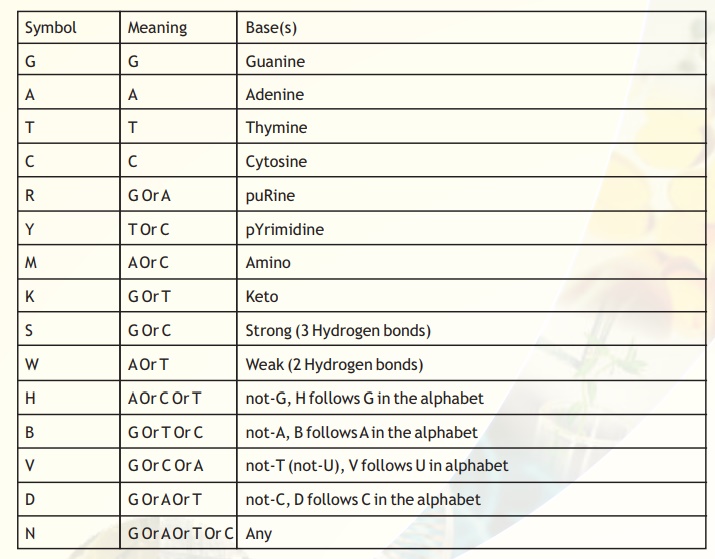
The symbols, their meaning and the bases for the nucleic acid sequences are presented in Table 3. The first 4 bases G,A,T,C, their symbols and the basis for nomenclature is clear. While determining sequence data through experiments, sometimes, the sequence identity at a particular position may not be clearly identifiable due to compression artifacts or other secondary structure related problems. In most cases the problem can be solved by repeating the experiment and also by sequencing the complementary strand. In a few cases, ambiguities may persist. In such cases, the most probable results are inferred from the chromatograms.
For instance, at a position where the ambiguity is not resolvable between a 'G' or a 'C' but one can be sure that there is no possibility of "A' or 'T' in the same position, then the symbol to be used is 'S'.
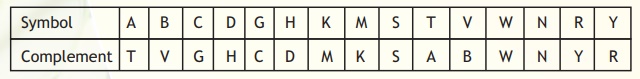
In most organisms, DNA is present as double stranded. The two strands are anti-parallel and complementary to each other (following Watson-Crick base-pairing). However, the problem arises when we start encountering the symbols that mean more than one base at a given position. Again, the IUPAC system comes to aid. The symbols to be used in the complementary strand corresponding to the symbol at the same position in a given strand are specified in Table 4. In certain cases, the complementary symbols are same as in the given strand because in both cases they mean the same set of bases.
Table 4. Definition of complementary symbols.
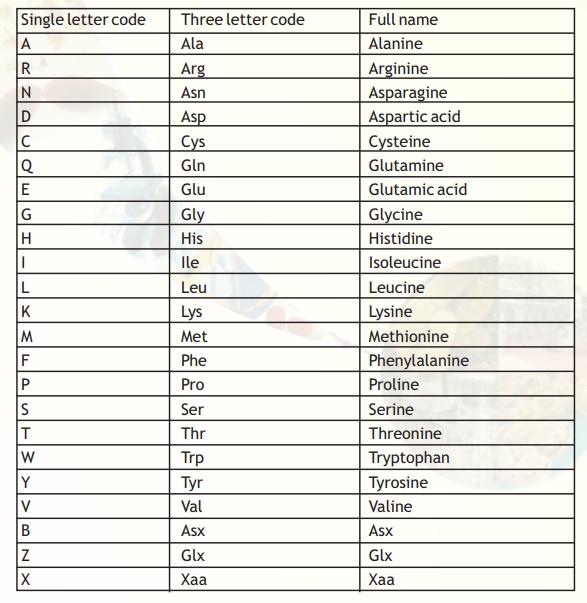
The symbols and their meaning for the protein sequences are presented in Table 5. It is evident that the number of symbols that mean more than one amino acid is very few.
Table 5. Symbol definitions for the amino acids.
The concept of directionality
In the biological systems, the usual direction in which the DNA and RNA are synthesized is the 5'-3' direction. This is universal and therefore it is helpful to adopt this fact as a way to collect and store data in the sequence databases. The nucleotide sequence are generally present in the database as they have been submitted or published, subject to some conventions which have been adopted for the database as a whole. The sequences are always listed in the direction 5' to 3'. Bases are numbered sequentially beginning with 1 at the 5' end of the sequence. The complementary sequence is described with a 'c' indicated next to the position of the sequence. Complementary sequence also runs 5'-3' but in the opposite direction to the given strand. Only one strand of the DNA sequence is given in a database entry. The complementary strand will have to be inferred using programs available in various packages or from various websites. We will see the details of a database entry in the next section. In the case of proteins, they are synthesized in the cell from N-terminus to the C-terminus. It is useful to adopt this convention in database entry for protein sequences. Thus, the concept of directionality used in biological systems is useful in describing the conventions to be adopted by the Database institutions. The advantage here is the universality of these fundamental biological processes in almost all living organisms.
Question : If you are given a sequence without any label, how will you find out whether it is a DNA sequence or a RNA sequence or a Protein sequence?
Answer : The usual approach taken by standard computer programs like sequence search programs scan the first 20 symbols. If the symbols encountered switch between any of the 4 bases only, then the sequence at hand is taken as a DNA sequence. Instead of T if U is encountered, then it is a RNA sequence. But if the symbols switch between any of the 20 (greater than 4), then it is taken as protein sequence.
Different types of sequences
cDNA : A large number of sequences deposited in the Databases were determined from cDNA molecules. While filling up the sequence entry form you must tick at the right position to indicate whether the sequence being deposited is a cDNA sequence. This data will also be provided when a sequence is retrieved. Thus in the case of cDNA sequences one is looking at the expressed part of the genome.
Genomic DNA: Sequencing of genomic DNA has become very routine nowadays. The genomic DNA is the store-house of information of which expressed part is represented in the cDNA sequences also.
ESTs : It is an abbreviation for Expressed Sequence Tags. Dr. Craig Venter initiated sequencing of a large number of cDNA molecules by sequencing one end of each of the randomly picked cDNA clones. Millions of ESTs have been deposited in a special database called dbEST. EST data is used to infer expression patterns by counting the number of ESTs corresponding to each gene divided by the total number of ESTs.
GSTs : In Plasmodium falciparum the enzyme Mung Bean Nuclease (MNase) cleaves in between the genes. A genomic DNA library generated by digestion with MNase was used for gene identification in P. falciparum. The approach used was similar to ESTs. One read of sequence was obtained from either ends. This data is referred to as genome sequence tags (GSTs). Usually, genomic DNA sequence refers to the nuclear DNA.
Organelle DNA: Eukaryotic cells have organelles such as mitochondria and chloroplast. These organelles have their own store house of information in the form of organelle DNA. Organelle DNA codes for a few genes. The coding information for the rest of the genes reside in the nuclear DNA of the same cell. If an organelle DNA has been sequenced the appropriate position in the sequence submission form must be mentioned.
Other molecules: In addition to these molecules, the databases contain the sequences of other molecules such as tRNA, and other small RNAs.
Related Topics