Chapter: Biotechnology: Genomics and Bioinformatics
Proteomics and Types of Proteomics
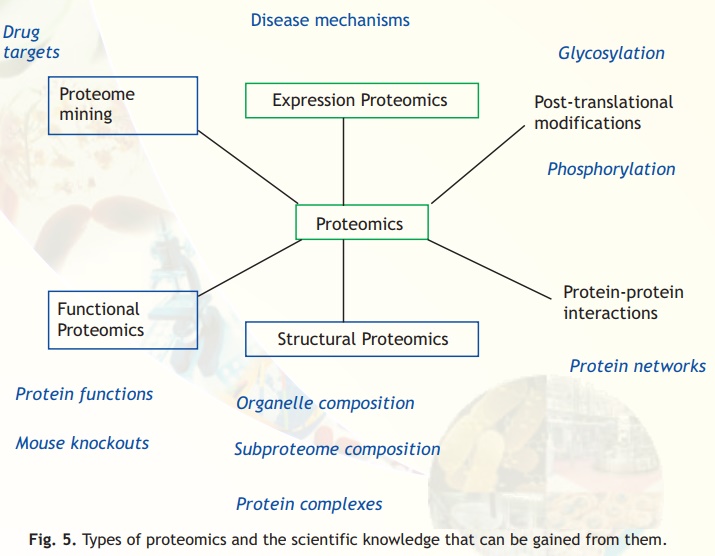
Proteomics
The term Proteome refers to the complete protein set of a cell. Proteomics refers to the large scale characterization of the entire protein complement of cells, tissues and even whole organisms. Modern proteomic studies involve many different areas, which are illustrated in Fig. 5. These include protein-protein interaction studies, protein function, and protein localization. The growth of proteomics is a direct result of advances made in large-scale nucleotide sequencing of various genomes. Without this development, protein identification would have been difficult.
It is important to have information about the proteins simply because they are responsible for the phenotype of the cells. It is impossible to understand mechanisms of disease, ageing etc. solely by studying the genome. Only by understanding protein function and their modifications, drug-targets for various diseases can be identified. One of the major aims of proteomics is to create a three dimensional map of a cell indicating the location of proteins.
The proteome of a given cell is dynamic. In response to internal and external cues biochemical machinery of the cell could be modulated. This could lead to several changes in the proteins such as post-translational modifications, changes in cellular localization, effect on their synthesis or degradation. Thus examination of a proteome is like taking a snapshot of the protein environment at a given time.
It is speculated that no function can be assigned to about one-third of the gene sequences in organisms for which the genome sequence is known. The complete identification of proteins in a genome will help structural genomics projects. The aim of these projects is to obtain 3D structure of all proteins in a genome. The structural analysis would be helpful in assigning function to many of these proteins. In addition to identification of proteins, one of the major goals of proteomics is to characterize post-translational modifications on proteins.
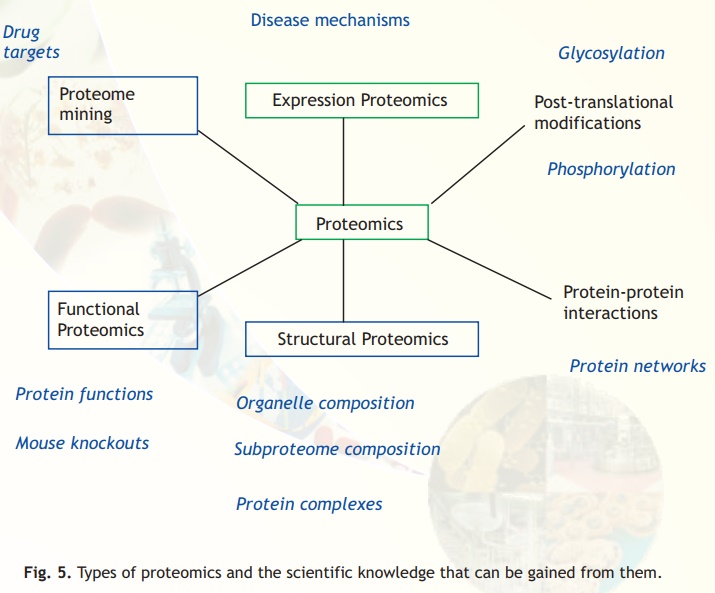
Fig. 5. Types of proteomics and the scientific knowledge that can be gained from them.
Types of Proteomics
Expression proteomics: The quantitative study of protein expression between samples that differ by some variable is known as expression proteomics. Using this approach, protein expression of the entire proteome or of subproteomes between samples can be compared. This could be useful in identification of disease specific proteins. For example: tumor samples from a cancer patient and a similar tissue sample from a normal individual could be analyzed for differential protein expression. Using two dimensional gel electrophoresis, followed by mass spectrometry, proteins, which are over or under expressed in the cancer patient compared to the normal individual can be identified. This could be compared with the microarray data (Fig. 4). Identification of these could provide a lead in understanding the basis of tumor development.
Structural proteomics: Unlike comparing the same cell or tissue in normal and diseased state in expression proteomics, structural proteomics are directed to map out the structure and nature of protein complexes present specifically in a particular cellular organelle. The aim is to identify all proteins present in a complex and to characterize all protein-protein interactions occurring between these proteins. Isolation of specific sub cellular organelles or protein complexes by purification can help assembling information about architecture of cells and explain how expression of certain proteins gives a cell its unique characteristics.
Functional proteomics: Functional proteomics is a very broad term for many specific, directed proteomics approaches. It can be defined as the use of proteomics methods to analyze the properties of molecular networks involved in a living cell. One of the major objectives is to identify molecules that participate in these networks. One of the successes of functional proteomics is identification and analysis of protein networks involved in the nuclear pore complex. This discovery has led to the identification of novel proteins which are important for translocating important molecules from the cytoplasm of a cell to the nucleus and vice versa.
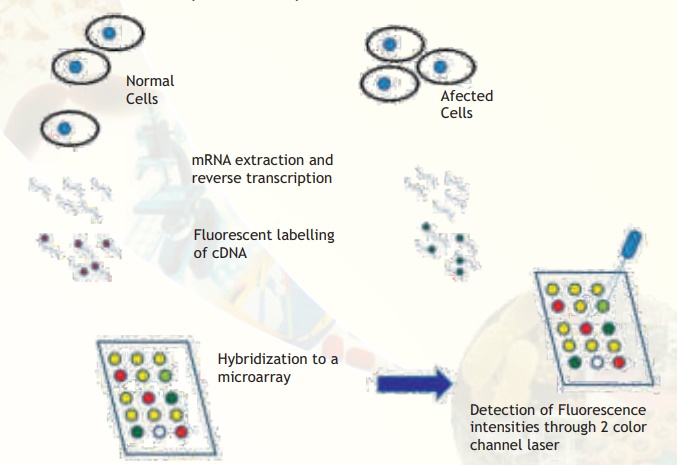
Fig. 4. Major steps involved in comparative microarray hybridization experiments between normal and affected (for example, cancerous) cells are illustrated. Observe that some spots are yellow, meaning that the particular gene is expressed in equal amounts. Some spots are clearly red or green indicating that the particular genes are expressed in only normal or affected condition. Some spots are more greenish or orange meaning that the expression status is not clearly tilted to either side but there is a trend towards either extremities. A few spots may appear blank (no colour).
Genes and Proteins
Number of genes vs Number of proteins
Analysis of mRNA does not provide a direct reflection of the protein content in the cell. One reason is that mRNA and protein expression levels do not always correlate. The formation of mRNA is only the first step in a long sequence of events resulting in protein synthesis (Fig. 6). mRNA could undergo various post-transcriptional modifications like, polyadenylation and mRNA editing. Some of these modifications could lead to the generation of various protein forms from a single gene. Subsequently, translational regulation of mRNA could take place. Proteins, after synthesis, could undergo post-translational modifications. It is estimated that proteins could undergo as many as 200 different types of these modifications. Due to these reasons the relationship between number of genes and number of proteins is not linear. In other words, the number of proteins could easily outnumber the number of genes.
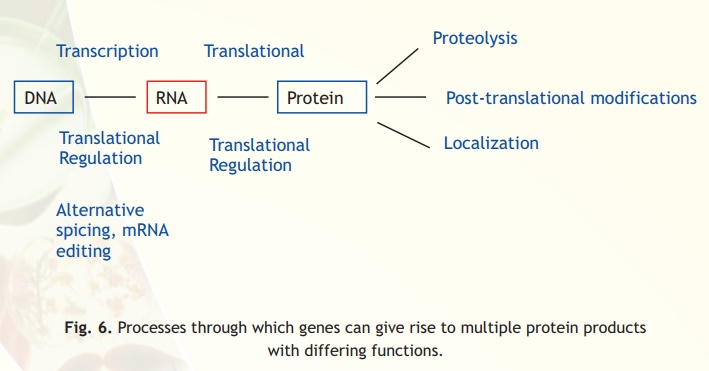
Fig. 6. Processes through which genes can give rise to multiple protein products with differing functions.
Related Topics