Chapter: Biotechnology: Genomics and Bioinformatics
Information Sources - Bioinformatics
Information Sources
Major databases
The National Center for Biotechnology Information (NCBI): The NCBI at the National Institutes of Health was created in 1988 to develop information systems in molecular biology. In addition to maintaining the GenBank nucleic acid sequence database, NCBI provides data retrieval systems and computational resources for the analysis of GenBank data and the variety of other biological data made available through NCBI.
The resources available from the NCBI have been classified into the following heads: (1) Database retrieval tools, (2) BLAST family of sequence similarity search programs, (3) Gene level sequences, (4) Chromosomal sequences, (5) Genome analysis, (6) Analysis of gene expression patterns, (7) Molecular structure.
All these Web based tools are available free. We shall learn about the practical aspects of some of these tools in practical classes. Below we discuss the first three set of resources. Learning these can help you in most cases. A great majority of bioinformatics activity is carried out using these resources. Other resources are useful for advanced studies.
Database retrieval tools
Among the database retrieval tools are ENTREZ, TAXONOMY BROWSER, LOCUS LINK. Entrez is an integrated database retrieval system. Through this system one can access literature (in the form of abstracts), sequences and structures. Entrez is an excellent system for obtaining comprehensive information on a given biological question. The taxonomy browser provides information on taxonomic classification of various species.
The taxonomy database has information on over 79, 000 organisms. Locus link carries information on the official gene names and other descriptive information about genes. Additionally, through Locus link one can access information on homologous genes. For example, it is very convenient to obtain information on the mouse homologue of a given human gene. Homologues from other organisms are also available.
BLAST family of search tools
Among the BLAST (Basic Local Alignment Search Tool) family of similarity search programs are several tools to analyze sequence information. These tools are designed to answer the question "Which sequences in the database are similar (or homologous) to my sequence?" The theory on which BLAST systems were developed is somewhat complex and is out of the scope of discussion here. The principles involved are-
(a) A given sequence is compared with sequences in the database using substitution matrices that specify scores to either 'reward' a match or 'penalize' a mismatch.
(b) Top scoring matches are ranked according to set criteria that serve to distinguish between a similarity due to ancestral relationship or due to random chance. In most analysis these criteria are not changed. However, if the user wishes, criteria can be changed.
(c) True matches are further examined thoroughly with other details accessible through Entrez and other tools available at NCBI.
Note: Two sequences being similar does not mean that they are homologous. Homology is defined as similarity due to common ancestry. Two sequences each from species A and species B are said to be homologous if they have descended from a common ancestor to species A and species B. Duplicated genes within a genome also may have similarities but these are referred to as 'paralogs'. Homologues will have the same function whereas paralogs may differ in functions.
Resources for gene level sequences
Among the resources for gene level sequences are several tools such as the UniGene, HomoloGene, RefSeq and others. We mentioned about the ESTs in the previous section. The method described therein produces many redundant ESTs because several cDNA clones represent the same gene. To manage the redundancy in EST data, UniGene database was created. The objective is to group ESTs into sets called clusters that belong to 'one' gene (Uni meaning one).
Homologene is a database of orthologs and homologs for several organisms like human, mouse, rat, zebrafish and cow genes represented in UniGene and Locus Link. It is easy to infer homologous relations using this database. RefSeq is a curated database of mRNAs and proteins of organisms like human, mouse and rat. The data provided in RefSeq has been used in many cases such as designing gene chips and describing the sequence features of the human genome.
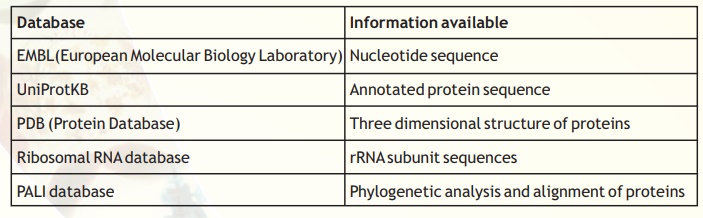
There are many other resources provided by the NCBI. Discussing all of these is not possible within the space limitations. Now, we mention a few other databases of importance to bioinformatics work (see Table 6 below)
Table 6. Examples of other useful databases for Bioinformatics.
Curator: A curator is one who reviews and checks newly submitted data ensuring all mandatory information has been provided, that biological features are adequately described and that the conceptual translations of any coding regions obey known translation rules. This process is called curation.
Related Topics