Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Molecular Biotechnology
Recombinant DNA Technology
RECOMBINANT DNA TECHNOLOGY
After it was established that DNA is the chemical constituent for the hereditary properties of the cell and after the discovery that (bacterial) cells can spontaneously take up DNA, investigators immedi-ately tried to manipulate the genetic properties of allkinds of cells. To achieve this, they simply added foreign DNA to microbial cells, plant cells or animal cells. All these attempts failed. There are two reasons for this lack of success. First of all, only a limited number of bacterial species is able to take up DNA spontaneously; most bacteria and certainly animal and plant species are unable to do this. Secondly, foreign DNA, if at all taken up, is in general not maintained in the receptor cell. DNA brought into a cell from outside will only be maintained if it is able to replicate autonomously, or if it is integrated in the recipient genome. In all other cases foreign DNA will not be propagated in the cell culture and will eventually disappear by degradation through the activity of cellular nucleases.
Genetic modification of organisms became fea-sible later on when recombinant DNA technologies were developed. This enabled the fusion of any DNA fragment to DNA molecules able to maintain them-selves by autonomous replication (such molecules are called replicons). The various specific techniques developed to introduce recombinant DNA molecules into all sorts of biological cells led to successful genetic modification strategies.
Replicons used as carriers for foreign DNA fragments are termed vectors. The vectors exploited in the DNA technology include mainly plasmids from bacteria or yeast, or DNA from bacterioviruses, animal viruses or plant viruses. Especially small microbial plasmids are very popular as vectors in biotechnology, since they can easily be isolated as intact circular double-stranded molecules. Plasmids with a broad host range, mentioned before, are very attractive since they can be used in various hosts and consequently enable a flexible application of the DNA technology.
For the application of plasmids in biotechnology, one has to fuse foreign DNA to the isolated plasmid in order to create a recombinant DNA molecule. The technology for this, the so-called recombinant DNA technology or DNA cloning technology, became feasible after the discovery of a specific class of nucleases, the so-called restriction endonucleases. Next to nucleases able to cleave any phosphodiester bond in DNA, nucleases which only cleave DNA at very specific sites are present in nature. These enzymes, the restriction enzymes, were discovered around 1970 in microorganisms. Their function is to discriminate between foreign DNA and self DNA. Microbial cells may in real life be confronted with DNA from an unrelated cell via various genetic transfer systems. Although all DNA is built likewise, DNA can be marked specifically at particular sites by a characteristic pattern of methylated or glucosylated nucleotides. This DNA marking, which does not interfere with the coding and replicating functions, is host specific. Restriction enzymes have the remark-able property to recognize DNA on the basis of the specific host marking. When DNA is transferred and the marking does not fit with the recipient cell, such DNA will be recognized and cut at specific sites by the restriction enzyme. Once the DNA is cut it will be further degraded in the cell. As is the case in many biological systems, things are never absolute: some DNA molecules may escape from the action of the restriction enzymes and by getting a proper marking they can be rescued.
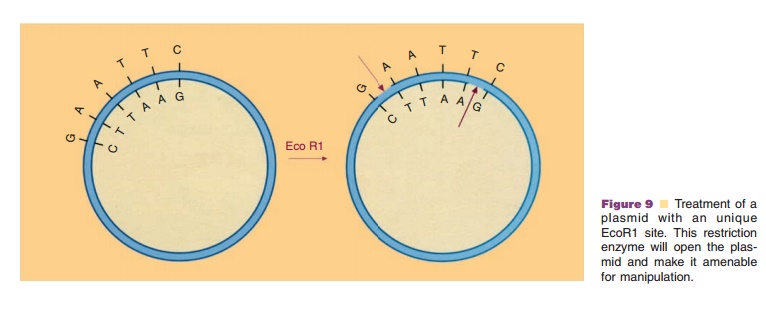
The very selective action of the restriction enzymes is the basis for their application in the recombinant DNA technology. Addition of a restric-tion enzyme to a plasmid without proper marking will convert the closed circular molecule to linear fragments, provided that the plasmid harbors recog-nition sites for the chosen restriction enzyme. In Figure 9 this is depicted for a special, but representa-tive case. A plasmid with only one recognition site for the restriction enzyme EcoR1 is treated with this enzyme. The double-stranded DNA is then asymme-trically cut at the recognition site, encompassing six bases namely GAATTC, which leads to linear DNA with typical short single-stranded ends. If foreign DNA—isolated either from microbial, plant or animal cells—with recognition sites for the enzyme EcoR1 is likewise cut with this enzyme, fragments with single-stranded ends characteristic for EcoR1 in the vector are formed. When the open vector and the foreign DNA fragments are brought together under appro-priate physicochemical conditions, the various single-stranded ends may recombine due to the presence of complementary bases. A possible reaction product could be a plasmid to which a specific foreign DNA fragment, as a passenger, is linked. Although the DNA pieces in such construct are interlinked by base pairing, they do not form a closed circular molecule. The enzyme DNA ligase, present in all sorts of cells and able to catalyze the formation of phosphodiester bonds, is used to create a closed circular recombinant DNA molecule. Although DNA fragments with complementary single-stranded ends (so-called cohe-sive ends) are the most favorable ones for linking, DNA ligase is also able to link fragments with blunt ends.
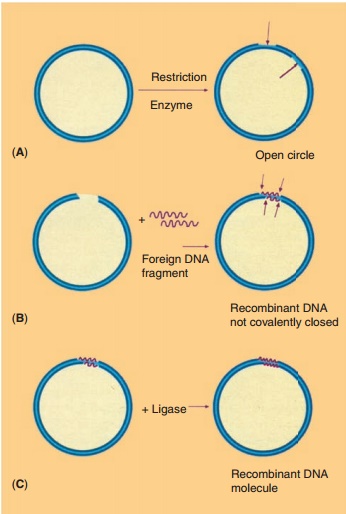
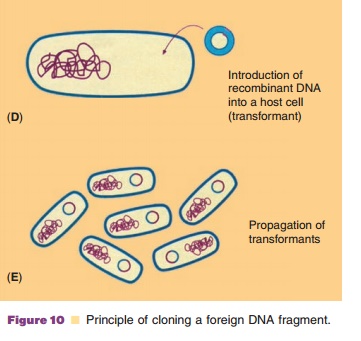
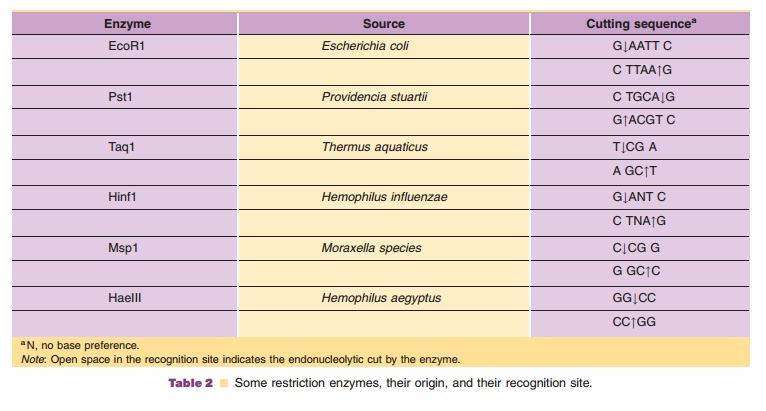
According to the technology presented schema-tically in Figure 10, recombinant DNA molecules consisting of vector and passenger DNA can be created. A great number of restriction enzymes with very specific recognition sites is available to cut DNA at a specific site. Some enzymes, like EcoR1, recognize a sequence of six base pairs, other enzymes recognize just four bases. Some, again likeEcoR1, cut the DNA asymmetrically while others create blunt ends when cutting DNA. In Table 2 some representative restric-tion enzymes are listed.
Recombinant DNA molecules are biologically of no interest as long as they reside in the reaction tube. Transfer of the construct to a living cell, however, may change the situation drastically. If the vector that served for the construct is able to replicate in the host, all daughter cells will inherit a precise copy (a clone) of the recombinant DNA molecule. Therefore the term “cloning” is frequently used for the technology described above.
The cloning technique is very suitable to obtain large amounts of a specific DNA fragment, by fusing such a fragment to an appropriate vector and transferring the construct to a host that can easily be cultivated to high cell densities. The recombinant DNA molecules, which can then be isolated from the cell mass, form an abundant source for the specific DNA fragment. Moreover and most important for pharmaceutical biotechnology, if the cloned piece of foreign DNA harbors an intact gene with appropriatesignals for gene expression, the modified host cells may, based on the universal character of gene expression, produce proteins encoded by the foreign DNA. This is the power of the recombinant DNA technology in a nutshell: an efficient way to amplify DNA fragments and a way to gain all sorts of gene products from hosts that one can choose.
Related Topics