Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Molecular Biotechnology
PCR Technology
PCR
Technology
For the detection of DNA or for testing the presence of mutations in DNA
the probe method described above is very powerful. Within the current probe
techniques, however, a substantial amount of DNA is required to allow the
detection of target DNA. The PCR (polymer-ase chain reaction) technology became
very popular in recent years to acquire large amounts of DNA.
In the PCR technology target DNA is amplified by in vitro DNA synthesis,
occurring in a number of fast repeating steps. The reaction starts with the
conversion of the double-stranded target DNA to single-stranded DNA and uses
specific oligonucleo-tides as primers to allow DNA polymerase to do its job.
The choice of the oligonucleotide primers, hybridizing with each of both target
strands, will determine the left and right limits of the DNA to be amplified.
Each PCR cycle (illustrated in Fig. 16) consists of three steps each
requiring only 1 to 3 minutes. In the first step the target DNA must be made
single stranded and this is done by heating the sample to 92LC. The second step involves the specific hybridiza-tion of the two
primers to the complementary single-stranded DNA. The optimal temperature for
this process is about 55LC. In the third step DNA
polymerase will extend the primer sequence using the single stranded DNA as a
template. The optimal extension temperature is about 72LC since the DNA polymerase chosen is derived from a thermophilic
bacterium, Thermus
aquaticus, which normally grows in hot
springs at temperatures above 80LC. This
DNA polymerase is extremely resistant against heat dena-turing and survives the
92LC DNA denaturing step. All reagents (target DNA, primers, dNTPs and
polymerase) are put in a tube which is sealed and usually 20 to 30 PCR cycles
are performed. The procedure can be automated and PCR machines are available
which control the temperature for each of the three separate steps of a PCR
cycle. Such machines can process hundreds of tubes simultaneously and produce
results within 2 to 4 hours.
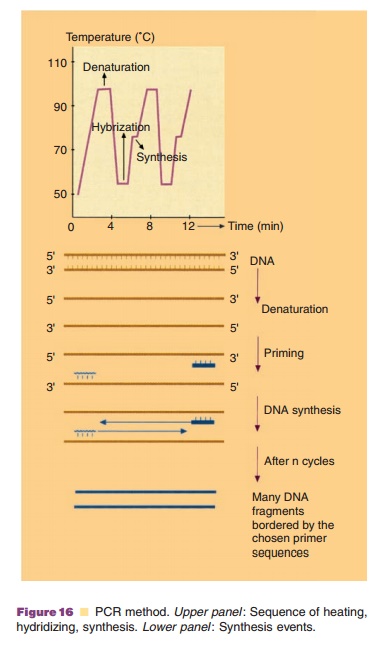
Ideally each cycle of DNA replication doubles the amount of DNA which is located in-between the chosen primers. Thirty PCR cycles will give an amplification of 230 times. This means that minute quantities of DNA can be amplified with specific primers to easily detectable levels. It should be realized that the specificity of the reaction is fully determined by the PCR primers and these primers will also determine the length of the amplified fragment. The tremendous sensitivity of the technique has sparked the development of a great number of applications where such sensitivity is of paramount importance. Also, compared to many other detection methods, the PCR procedure is very fast.
For example, the presence of microbial patho-gens in raw and processed
food products can be unequivocally determined using this technology. DNA is
extracted from this material and the PCR reaction is performed using primers
which are specific for the suspected pathogen(s). Detailed knowledge of DNA
sequences of all sorts of genes in all sorts of organisms allows the
development of such specific primers, the main prerequisite for diagnostic PCR
technology. If specific amplified DNA products can be detected, this is proof
that the pathogen is present in the material. Also in clinical material (blood,
urine, etc.) the technique is usedextensively as a rapid and sensitive test for
the presence of bacterial and viral pathogens. A third area where PCR has
become standard technology is in forensic science. At a crime scene often
minute quantities of potentially important evidence is found (single hairs,
blood drops, semen stains, etc.) and PCR technology can be used to get enough
DNA to show the origin of this material. These are only a few examples of the
use of PCR technology. PCR is often an essential step in elaborate diagnostic
and detection procedures and novel applications are continuously being
developed.
As for the application of the PCR technology for diagnosis of pathogens,
one has to realize that for most purposes the intent is to detect viable
pathogens. The PCR technology obviously cannot distinguish DNA from vital or
dead material and in that respect it is not always an adequate technique. The
PCR technique is a very sensitive one since minute amounts of DNA are highly
amplified. This high sensitivity may limit the discriminative power of the
technique when applied for diagnostic purposes. For example, it may detect
minor contaminants in the samples. Moreover, DNA contaminants may be introduced
during the perfor-mance of the tests. It is therefore a major concern in the
application of PCR to avoid DNA contaminations that could cause false positive
reactions.
Modified PCR techniques and related methodol-ogies are discussed.
Related Topics