Chapter: Essential Microbiology: Biochemical Principles
Proteins - Biomacromolecules
Proteins
Of the macromolecules commonly found in living systems, proteins are the most ver-satile, having a wide range of biological functions and this fact is reflected in their structural diversity.
The five elements found in most naturally occurring proteins are carbon, hydrogen, oxygen, nitrogen and sulphur. In addition, other elements may be essential components of certain specialised proteins such as haemoglobin (iron) and casein (phosphorus).
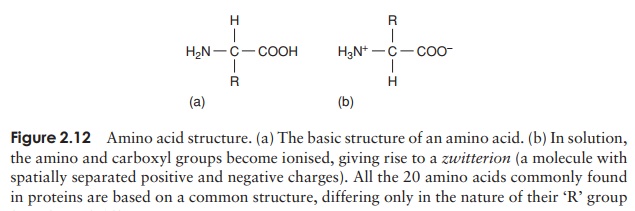
Proteins can be very large molecules, with molecular weights of tens or hundreds of thousands. Whatever their size, and in spite of the diversity referred to above, all proteins are made up of a collection of ‘building bricks’ called amino acids joined together. Amino acids are thought to have been among the first organic molecules formed in the early history of the Earth, and many different types exist in nature. All these, including the 20 commonly found occurring in proteins, are based on a common structure, shown in Figure 2.12. It comprises a central carbon atom (known as the α-carbon) covalently bonded to an amino (NH2) group, a carboxyl (COOH) group and a hydrogen atom. It is the group attached to the final valency bond of the α-carbon which varies from one amino acid to another; this is known as the ‘R’-group.
The 20 amino acids found in proteins can be conveniently divided into five groups, on the basis of the chemical nature of their ‘R’-group. These range from a single hydrogen atom to a variety of quite complex side chains (Figure 2.13). It is unlikely nowadays that you would need to memorise the precise structure of all 20, as the author was asked to do in days gone by, but it would be advisable to familiarise yourself with the groupings and examples from each of them. The groups differentiate on the basis of a polar/non-polar nature and on the presence or absence of an ionisable ‘R’-group.
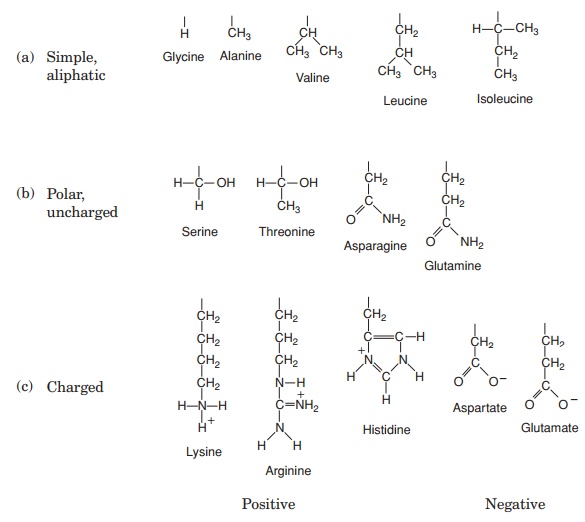
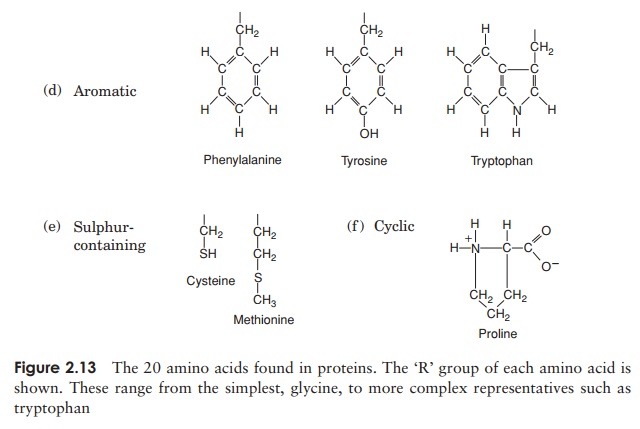
Note that one amino acid, proline, falls outside the main groups. This differs from the others in that it has one of its N—H linkages replaced by an N—C, which forms part of a cyclic structure (Figure 2.13). This puts certain conformational constraints upon proteins containing proline residues.
As can be seen from Figure 2.13, the simplest amino acid is glycine, whose R-group is simply a hydrogen atom. This means that the glycine molecule is symmetrical, with a hydrogen atom on opposite valency bonds. All the other amino acids however, are asymmetrical. The α-carbon acts as what is known as a chiral centre, giving the molecule right or left ‘handedness’. Thus two stereoisomers known as the D- and L-forms are possible for each of the amino acids except glycine. All the amino acids found in naturally occurring proteins have the L-form; the D-form also occurs in nature but only in certain specific, non-protein contexts.
Proteins, as we’ve seen, are polymers of amino acids. Amino acids are joined together by means of a peptide bond. This involves the -NH2 group of one amino acid and the -COOH group of another. The formation of a peptide bind is a form of condensation reaction in which water is lost (Figure 2.14).
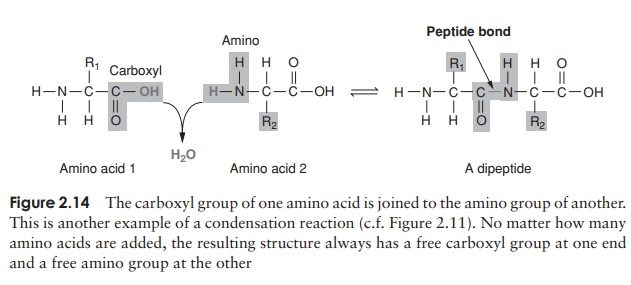
The resulting structure of two linked amino acids is called a dipeptide; note that this structure still retains an -NH2 at one end and a -COOH at the other. If we were to add on another amino acid to form a tripeptide, this would still be so, and if we kept on adding them until we had a polypeptide, we would still have the same two groupings at the extremities of the molecule. These are referred to as the N-terminus and the C-terminus of the polypeptide. Since a water molecule has been removed at the formation of each peptide bond, we refer to the chain so formed as being composed of amino acid residues, rather than amino acids. The actual distinction between a protein and a polypeptide based on the number of amino acid residues is not clear-cut; generally, with over 100, we refer to proteins, but some naturally occurring proteins are a lot smaller than this.
So far, we can think of proteins as long chains of many amino acid residues, rather like a string of beads. This is called the primary structure of the protein; it is deter-mined by the relative proportions of each of the 20 amino acids, and the order in which they are joined together. It is the basis of all the remaining levels of structural complexity, and it ultimately determines the properties of a particular protein. It is also what makes one pro-tein different from another. Since the 20 types of amino acid can be linked together in any order, the number of possible sequences is astronomical, and it is this great variety of structural possibilities that gives proteins such diverse structures and functions.
Some parts of the primary sequence are more important than others. If we took a protein of, say, 200 amino acid residues in length, took it apart and reassembled the amino acids in a different order, we would almost certainly alter (and probably lose completely) the properties of that protein. If we look at the primary sequence of a protein molecule which serves essentially the same function in several species, we find that nature has allowed slight alterations to occur during evolution, but these are often conservative substitutions, where an amino acid has been replaced by a similar one (one from the same group in Figure 2.13), and thus have little effect on the pro-tein’s properties. In certain parts of the primary sequence, such substitutions are less well tolerated, for example the few residues that make up the active site of an enzyme. In cases such as the one above, alterations have not been allowed at these points in the primary sequence, and the sequence is the same, or almost so, in all species possessing that protein. The sequence in question is said to have been conserved.
Higher levels of protein structure
The structure of proteins is a good deal more complicated than a just a linear chain of amino acids. A long thin chain is unlikely to be very stable; proteins therefore undergo a process of folding which makes the molecule more stable and compact. The results of this folding are the secondary and tertiary structures of a protein
The secondary structure is due to hydrogen bonding between a carbonyl (-CO) group and an amido (-NH) group of amino acid residues on the peptide backbone (Figure 2.15). The ‘R’ group plays no part in secondary protein structure. Two regular patterns of folding result from this; the α-helix and the β-pleated sheet.
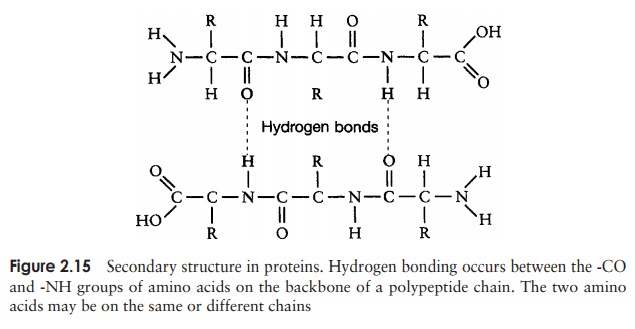
The α-helix occurs when hydrogen bonding takes place between amino acids close together in the primary structure. A stable helix is formed by the -NH group of an amino acid bonding to the -CO group of the amino acid four residues further along the chain (Figure 2.16a). This causes the chain to twist into the characteristic he-lical shape. One turn of the helix occurs every 3.6 amino acid residues, and results in a rise of 5.4 A (0.54 nm); this is called the pitch height of the helix. The ability to form a helix like this is dependent on the component amino acids; if there are too many with large R-groups, or R-groups carrying the same charge, a stable helix will not be formed. Because of its rigid structure, proline (Figure 2.13) cannot be accommodated in anα-helix. Naturally occurring α-helices are always right-handed, that is, the chain of amino acids coils round the central axis in a clockwise direction. This is a much more stable configuration than a left-handed helix, due to the fact that there is less steric hindrance (overlapping of electron clouds) between the R-groups and the C==O group on the peptide backbone. Note that if proteins were made up of the D-form of amino acids, we would have the reverse situation, with a left-handed form favoured. In the β-pleated sheet, the hydrogen bonding occurs between amino acids either on separate polypeptide chains or on residues far apart in the primary structure (Figure 2.16b). The chains in a β-pleated sheet are fully extended, with 3.5 A (0.35 nm) between adjacen amino acid residues (c.f. α-helix, 1.5 A). When two or more of these chains lie next to each other, extensive hydrogen bonding occurs between the chains. Adjacent strands in a β-pleated sheet can either run in the same direction (e.g. N→C), giving rise to a par-allel β-pleated sheet, or in opposite directions (antiparallel β-pleated sheet, as shown in Figure 2.16b).
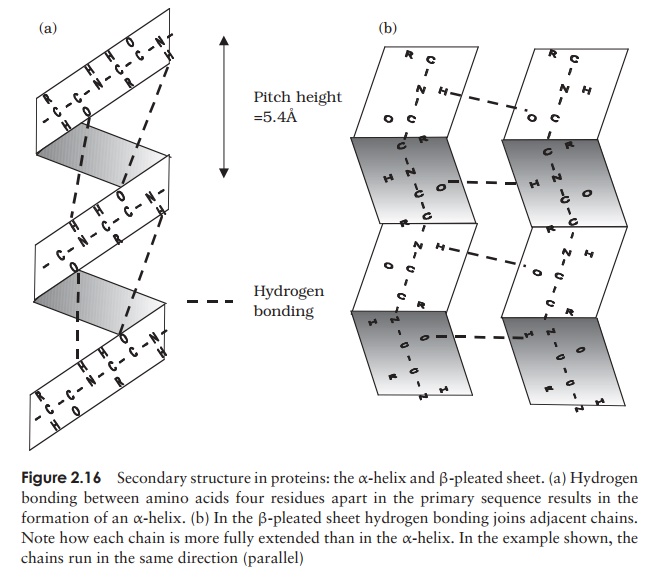
A common structural element in the secondary structure of proteins is the β-turn. This occurs when a chain doubles back on itself, such as in an antiparallel β-pleated sheet. The -CO group of one amino acid is hydrogen bonded to the -NH group of theresidue three further along the chain. Frequently, it is called a hairpin turn, for obvious reasons (Figure 2.17). Numerous changes in direction of the polypeptide chains result in a compact, globular shape to the molecule.
Typically about 50 per cent of a protein’s secondary structure will have an irregular form. Although this is often referred to as random coiling, it is only random in the sense that there is no regular pattern; it still contributes towards the stability of the molecule. The proportions and combinations in which α-helix, β-pleated sheet and random coiling occur varies from one protein to another. Keratin, a structural protein found in skin, horn and feathers, is an example of a protein entirely made up of α-helix, whilst the lectin (sugar-binding protein) concanavalin A is mostly made up of β-pleated sheets.
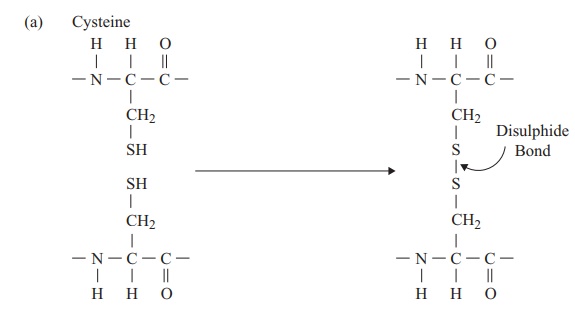
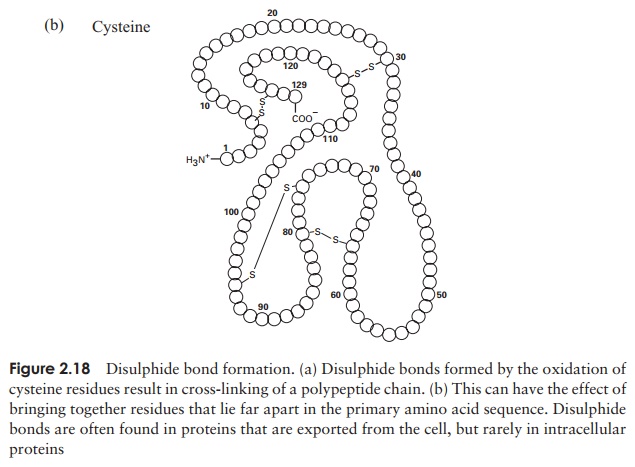
The tertiary structure of a protein is due to interactions between side chains, that is, R-groups of amino acid residues, resulting in the folding of the molecule to produce a thermodynamically more favourable structure. The structure is formed by a variety of weak, non-covalent forces; these include hydrogen bonding, ionic bonds, hydrophobic interactions, and Van der Waals forces. The strength of these forces diminishes with distance, therefore the formation of a compact structure is encouraged. In addition, the -SH groups on separate cysteine residues can form a covalent -S—S- linkage. This is known as a disulphide bridge and may have the effect of bringing together two cysteine residues that were far apart in the primary sequence (Figure 2.18).
In globular proteins, the R-groups are distributed according to their polarities; non-polar residues such as valine and leucine nearly always occur on the inside, away from the aqueous phase, while charged, polar residues including glutamic acid and histidinegenerally occur at the surface, in contact with the water.
The protein can be denatured by heating or treatment with certain chemicals; this causes the tertiary structure to break down and the molecule to unfold, resulting in a loss of the protein’s biological properties. Cooling, or removal of the chemical agents, will lead to a restora-tion of both the tertiary structure and biological activity, showing that both are entirely dependent on the primary sequence of amino acids.
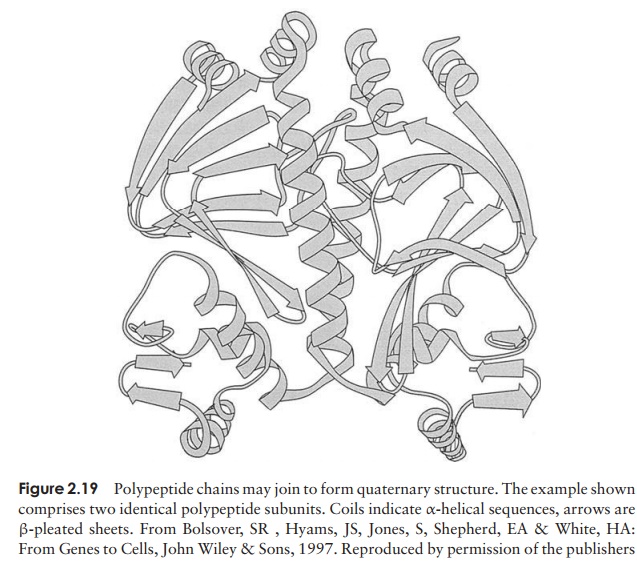
Even the tertiary structure is not always the last levelof organisation of a protein, because some are made up of two or more polypeptide chains, each with its own secondary and tertiary structure, combined together to give the quaternary structure (Figure 2.19). These chains may be identical or different, dependingon the protein. Like the tertiary structure, non-covalent forces between R-groups are responsible, the difference being that this time they link amino acid residues on separate chains rather than on the same one.
Such proteins lose their functional properties if dissociated into their constituent units; the quaternary joining is essential for their activity. Phosphorylase A, an enzyme involved in carbohydrate metabolism, is an example of a protein with a quaternary structure. It has four subunits, which have no catalytic activity unless joined together as a tetramer.
Although all proteins are polymers of amino acids existing in various levels of structural complexity as we have seen above, some have additional, non-amino acid components. They may be organic, such as sugars (gly-coproteins) or lipids (lipoproteins) or inorganic, includ-ing metals (metalloproteins) or phosphate groups (phos-phoproteins). These components, which form an integral part of the protein’s structure, are called prosthetic groups.
Related Topics