Chapter: Basic Concept of Biotechnology : Biotechnology in Forensic Sciences
Polymerase Chain Reaction (PCR) - Biotechnology in Forensic Sciences
Polymerase Chain Reaction (PCR):
During the investigation of a crime, the amount of DNA evidence procured is often very small, thus for efficient DNA profiling, amplification is ideal. PCR allows for the exponential amplification of DNA fragments to the lengths of approximately 10,000bp. PCR is particularly helpful in the amplification of small amounts or degraded samples. In the 1980s, when RFLP was being developed for forensic use, PCR was developed for forensic applications. In 1983, a biochemist, Dr Kary Mullis adopted PCR to amplify DNA fragments of forensic interest in an automated process. He was awarded the Nobel Prize for his discoveryin 1993. PCR solved one of the major struggles facing by forensic biologists. RFLP typing required relatively large quantities of DNA.
The PCR Process
PCR methods are very susceptible to contamination by foreign DNA. On this ground, DNA extractions are always performed in a location physically isolated from the desk/lab where the subsequent amplifications would be performed.
A PCR reaction needs a number of fundamental primary components. Oligonucleotide primers complementary to the DNA target to mark the target to be amplified, with two primers in use. Fluorescent tags are sometimes added to the primers to locate amplified DNA in electrophoresis. DNA polymerase enzyme adds nucleotides to the 3’ end of the primers and allows the DNA strand to be copied. Other components required include a template DNA, a reaction buffer with MgCl2 to ascertain ideal conditions for the performance of the DNA polymerase enzyme and deoxyribonucleotides to build the DNA molecule.
The three steps of PCR process take place within a thermal cycler, capable of reaching and upholding preset temperatures very accurately. The DNA samples are supplemented to a reaction buffer, a salt solution that is buffered at the optimal pH so that the polymerase enzyme can function properly. The building blocks of DNA, four nucleotides, are added to the buffer along with the Taq polymerase enzyme that helps in catalysing the extension step.
The steps in the PCR reaction are as follows:
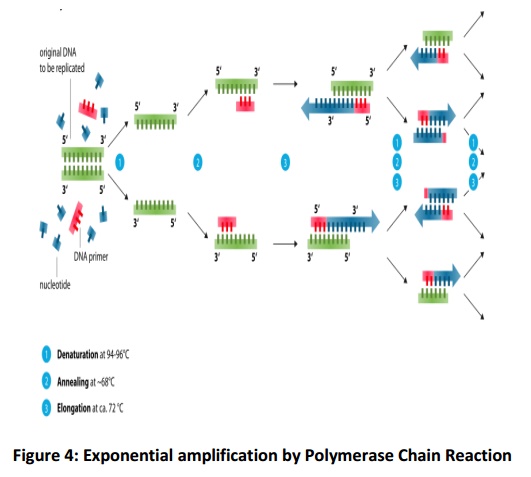
Denaturation
After adding the DNA to the PCR tube containing the reaction mixture it is heated to 95°C which leads to the denaturation of the double-stranded DNA. The H-bonds present between the base pairs break and single-stranded DNA is formed. Each strand becomes the template for the formation of a new double-stranded DNA.
Annealing
In this step, a short strand of synthetic DNA, primer, attaches to each of the separated strands. They mark the starting points for the addition of new bases to start the replication of each strand. The temperature of thermal cycler drops to 60°C for this step.
Extension
Under the influence of Taq polymerase at the temperature of 72°C, single bases/ nucleotides are added to the primer, one by one. At each of the complementary single strands created by the denaturation process, this process occurs so at the end two identical pieces of double-stranded DNA are produced. This makes one PCR cycle. The temperature is raised once again to 94°C and the process repeats in this way .Four single strands are formed after the denaturation of the two strands. They again go through annealing and extension, which forms four new double strands. The process continues till 25–40 cycles, which takes nearly 2-3 hours. This produces approximately one billion copies of the original DNA, sufficient for additional typing. The steps in PCR are shown in Fig.4.
Basic PCR Protocol
The protocol serves as a wide-ranging guideline and a preliminary point for any PCR amplification. Optimal reaction conditions (primers, MgCl2, incubation times and temperatures, concentration of Taq DNA Polymerase and template DNA) may differ and need to be calculatedfor the experiment.
1. Add the following components to a DNase/RNase-free 0.5-ml microcentrifuge tube placed once. Scale the reaction volumes as needed. Prepare a master mix for multiple reactions, to minimize reagent loss and to enable accurate pipetting.
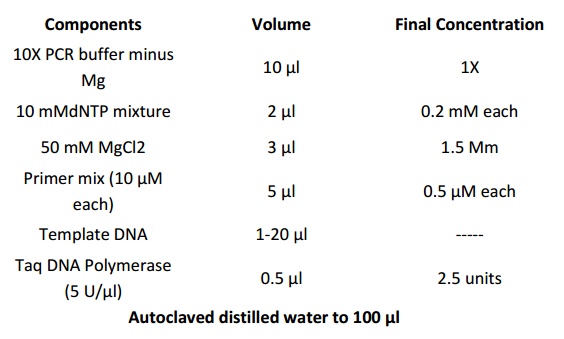
2. Mix contents of tube and overlay with 50 µl of mineral or silicone oil.
3. Cap tubes and centrifuge briefly to collect the contents to the bottom.
Before the amplification of DNA in a thermal cycler, the DNA must be extracted from the collected sample from crime scene. Several steps need to be taken to isolate DNA from the cells, the cell membrane and nucleus are broken to expose the DNA without destroying it. This can be achieved through numerous different extraction methods. Chelex, created by the Bio-Rad company, is a resin that is added to a sample of DNA. Chelex resins being negatively charged help to remove positive metal ions. Chelex resins bind Mg+ ions in order to prevent DNA nucleases from becoming activated. After the centrifugation of the sample, purified DNA can be removed from the supernatant.
Other methods of extracting DNA include specialized cellulose paper called FTA™, the use of organic chemicals (phenol-chloroform) and differential extraction. All achieve the same purpose.
DNA Typing of PCR Product
The products aredetected after the amplification process is complete. Since the product is more relative to that produced by RFLP, it is not compulsory to employ very delicate detection methods such as radioactive labelling. A yield gel experiment can be performed on agarose and stain the product with EtBr.
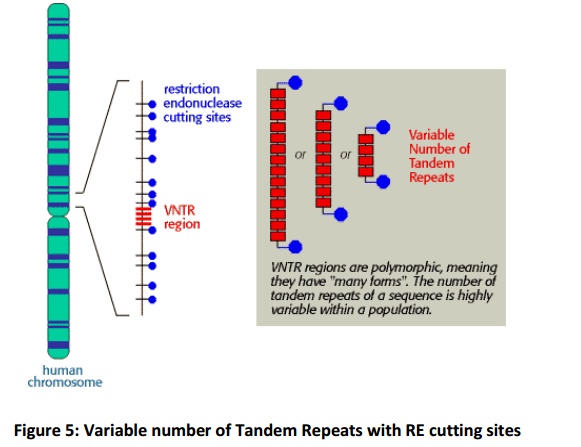
Variable Number of Tandem Repeats (VNTRs)
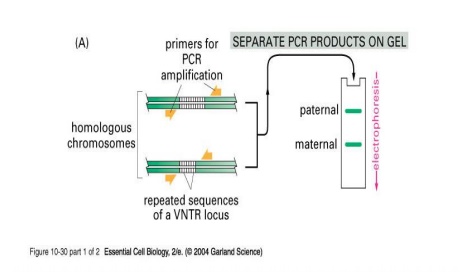
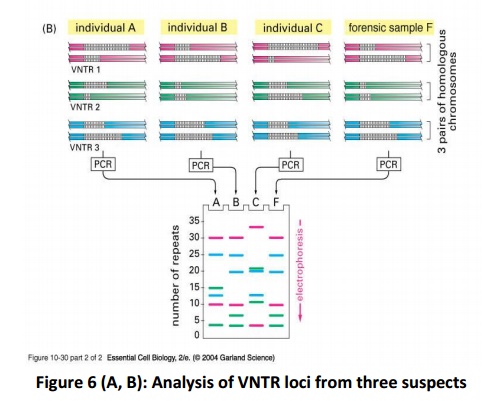
The DNA sequences that create the variability contain runs of short, repeated sequences, such as GTGTGT..., which are presentin various positions (loci) in the human genome. The number of repeats in each run is extremely variable in any population, ranging from 4 to 40 in different individuals. This type of segment of repeated nucleotides is generally referred to as a hypervariablemicrosatellite sequence, also known as a VNTR (variable number of tandem repeats) sequence (Fig.5). Individuals usually inherit a different variant of each VNTR locus from their mother and from their father because of the variability in these sequences; two unrelated individuals therefore do not generally contain the same pair of sequences. A PCR reaction using primers produces a pair of bands of amplified DNA from each individual, one band representing the paternal variant and the other representing the maternal variant(Fig.6A). The length of the amplified DNA and its position after electrophoresis depend on the exact number of repeats at the locus.
In the schematic example shown in Fig.6B, the three VNTR loci are analyzed from three suspects (individuals A, B, and C), producing six bands for each person A,B and C after polyacrylamide gel electrophoresis. Nevertheless, some individuals have numerous bands in common; the overall pattern is quite unique for each. The band pattern can therefore serve as a fingerprint to identify an individual nearly uniquely. The fourth lane, F, contains the products of the same PCR reactions carried out on the forensic sample. The starting material for such a PCR reaction can be a single hair, scratched tissue or a tiny sample of blood that was left at the crime scene. The more loci that are examined, the more confident one can be about the results. While examining the variability at 5-10 different VNTR loci, the odds that two random individuals would share the same fingerprint by chance are approximately one in 10 billion. In the case shown here, individuals A and C can be eliminated from inquiries, while B remains a clear suspect. A similar approach is now used routinely for paternity testing.
The application of PCR technology to forensic science
For the PCR to be a useful application in forensic science the selected target sequences should carry several allelic variations, possibly all less than 1000 bp in size. A range of polymorphic loci that fits for biological discrimination purposes fulfils these parameters. The best known of the PCR systems used for forensic science are the D1S80 and the HLA DQA1systems.
The HLA DQA1 kit uses a series of allele specific probes to identify the various allelic forms.
D1S80 is a VNTR locus where the allelic forms are simply separated and identified by size. The term AMP-FLP's has been used to define D1S80 and other VNTR loci that can be amplified by PCR. Other AMP-FLP's that have also proved to be useful PCR systems are the 3' HVR (Hyper variable repeats) region of the apolipoprotein B gene, D17S30 and Col1A2.
A third PCR system, the mitochondrial D loop region, has more recently fascinated a deal of attention by forensic scientists. This is a highly polymorphic DNA locus present on the mitochondrial genome. When the D loop locus is once amplified, it is best studied by direct sequencing of the PCR products. This system is likely to receive a great deal ofinterest with the aid of modern automated DNA sequences.
Advantages of PCR technology
1. It is more sensitive than the single-locus and multi-locus probe technologies and the protein and antigenic systems.
2. The PCR process can be tailored for a particular locus.
3. The technology is potentially very cheap.
4. The technology is simple to understand and much easier to perform than RFLP technology.
5. Radioactive isotopes are not required.
6. Results can often be obtained from crude DNA preparations.
7. The PCR products do not alter by degradation of the template DNA caused by decomposition of the sample.
8. The amplification process is largely automated, and therefore precise.
Disadvantages of PCR technology
1. False positives may occur if care is not taken to prevent the contamination of samples.
2. The PCR process may be inhibited by various chemicals present in the DNA extract viz.Haemoglobin, metabolites released by vaginal flora and various organic acids in soils are powerful inhibitors of the PCR process.
3. PCR errors like inability of proof reading of Taq DNA polymerase may result in error in amplification. Mispriming and excessive primer dimer formation may also occur.
Related Topics