Chapter: Basic & Clinical Pharmacology : Drugs Used in Asthma
Pathogenesis of Asthma
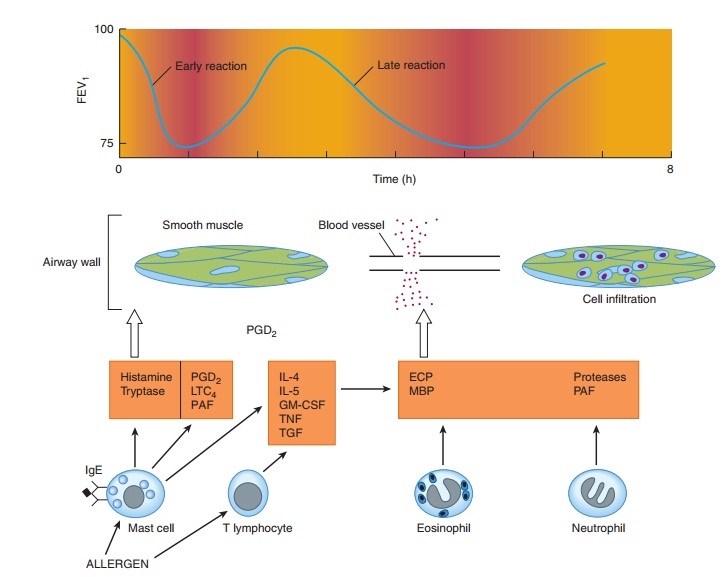
PATHOGENESIS OF ASTHMA
The
classic immunologic model of asthma presents it as a disease mediated by
reaginic immune globulin (IgE). Foreign materials that provoke IgE production
are described as “allergens”; the most common are proteins from house dust
mite, cockroach, animal danders, molds, and pollens. The tendency to produce
IgE antibodies is genetically determined; asthma and other aller-gic diseases
cluster in families. Once produced, IgE antibodies bind to mast cells in the
airway mucosa (Figure 20–1). On reexposure to a specific allergen,
antigen-antibody interaction on the surface of the mast cells triggers both the
release of mediators stored in the cells’ granules and the synthesis and
release of other mediators. The histamine, tryptase, leukotrienes C 4
and D4, and prostaglandin D2 that are released diffuse
through the airway mucosa, triggering the muscle contraction and vascular
leakage responsible for the acute bronchoconstriction of the “early asth-matic
response.” This response is often followed in 3–6 hours by
a
second, more sustained phase of bronchoconstriction, the “late asthmatic
response,” which is associated with an influx of inflam-matory cells into the
bronchial mucosa and with an increase in bronchial reactivity that may last for
several weeks after a single inhalation of allergen. The mediators responsible
for this late response are thought to be cytokines characteristically produced
by TH2 lymphocytes, especially interleukins 5, 9, and 13. These cytokines are
thought to attract and activate eosinophils, stimu-late IgE production by B
lymphocytes, and stimulate mucus production by bronchial epithelial cells. It
is not clear whether lymphocytes or mast cells in the airway mucosa are the
primary source of the mediators responsible for the late inflammatory response,
but the benefits of corticosteroid therapy are attributed to their inhibition
of the production of pro-inflammatory cyto-kines in the airways.
The
allergen challenge model does not reproduce all the fea-tures of asthma. Most
asthma attacks are not triggered by inhala-tion of allergens. They are
triggered by viral respiratory infection. Some adults with asthma have no
evidence of allergic sensitivity to allergens, and even in people with allergic
sensitivity, the severity of symptoms correlates poorly with levels of allergen
in the atmo-sphere. Moreover, bronchospasm can be provoked by nonaller-genic
stimuli such as distilled water, exercise, cold air, sulfur dioxide, and rapid
respiratory maneuvers.
This
tendency to develop bronchospasm on encountering stimuli that do not affect
healthy nonasthmatic airways is charac-teristic of asthma and is sometimes
called “nonspecific bronchial hyperreactivity” to distinguish it from bronchial
responsiveness to specific antigens. Bronchial reactivity is assessed by
measuring the fall in forced expiratory volume in 1 second (FEV1)
provoked by inhaling serially increasing concentrations of aerosolized
metha-choline. The exaggerated reactivity of the airways appears to be fundamental
to asthma’s pathogenesis, because it is nearly ubiqui-tous in patients with
asthma and its degree roughly correlates with the clinical severity of the
disease.
The
mechanisms underlying bronchial hyperreactivity are somehow related to
inflammation of the airway mucosa. The agents that increase bronchial
reactivity, such as ozone exposure, allergen inhalation, and infection with
respiratory viruses, also cause airway inflammation. The increase in reactivity
due to aller-gen inhalation is associated with an increase in both eosinophils
and polymorphonuclear leukocytes in bronchial lavage fluid. The increase in
reactivity that is associated with the late asthmatic response to allergen
inhalation (Figure 20–1) is sustained and, because it is prevented by treatment
with an inhaled corticoster-oid, is thought to be caused by airway
inflammation.
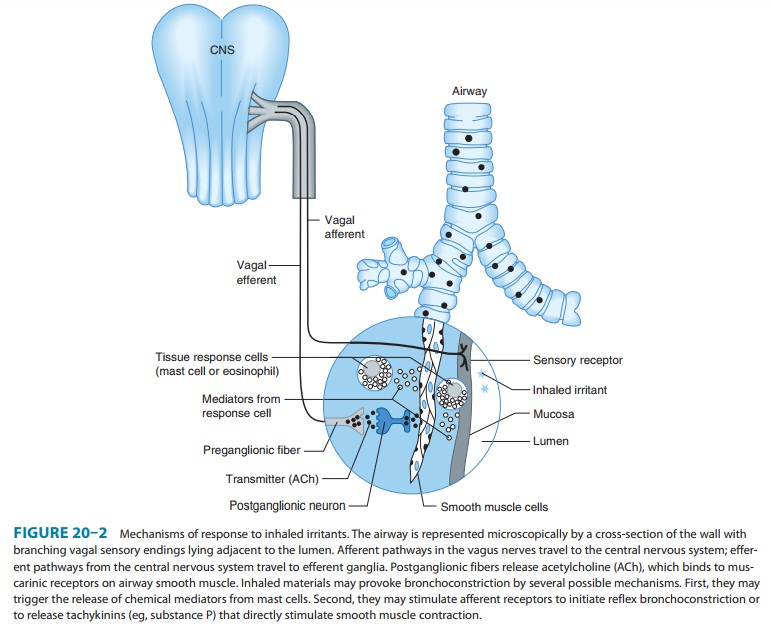
Whatever
the mechanisms responsible for bronchial hyperreac-tivity, bronchoconstriction
itself seems to result not simply from the direct effect of the released mediators
but also from their acti-vation of neural or humoral pathways. Evidence for the
impor-tance of neural pathways stems largely from studies of laboratory
animals. The bronchospasm provoked in dogs by inhalation of histamine is
reduced by pretreatment with an inhaled topical anesthetic agent, by
transection of the vagus nerves, and by pre-treatment with atropine. Studies of
asthmatic humans, however, have shown that treatment with atropine causes only
a reduction
in—not
abolition of—the bronchospastic responses to antigens and to nonantigenic
stimuli. It is possible that activity in another neural pathway, such as the
nonadrenergic, noncholinergic sys-tem, contributes to bronchomotor responses to
stimuli (Figure 20–2).
The
hypothesis suggested by these studies—that asthmatic bronchospasm results from
a combination of release of media-tors and an exaggeration of responsiveness to
their effects— predicts that asthma may be effectively treated by drugs with
different modes of action. Asthmatic bronchospasm might be reversed or
prevented, for example, by drugs that reduce the amount of IgE bound to mast
cells (anti-IgE antibody), pre-vent mast cell degranulation (cromolyn or
nedocromil, sym-pathomimetic agents, calcium channel blockers), block the
action of the products released (antihistamines and leukotriene receptor
antagonists), inhibit the effect of acetylcholine released from vagal motor
nerves (muscarinic antagonists), or directly relax airway smooth muscle
(sympathomimetic agents, theophylline).
The
second approach to the treatment of asthma is aimed not only at preventing or
reversing acute bronchospasm but at reducing the level of bronchial
responsiveness. Because increased responsive-ness appears to be linked to
airway inflammation and because air-way inflammation is a feature of late
asthmatic responses, this strategy is implemented both by reducing exposure to
the allergens that provoke inflammation and by prolonged therapy with
anti-inflammatory agents, especially inhaled corticosteroids.
Related Topics