Chapter: Basic & Clinical Pharmacology : Beta-Lactam & OtherCell Wall- & Membrane-Active Antibiotics
Other Cell Wall or Membrane Active Agents
OTHER CELL WALL OR MEMBRANE ACTIVE AGENTS
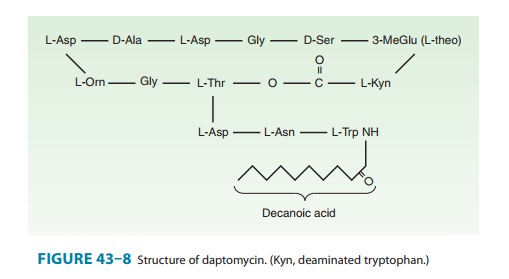
DAPTOMYCIN
Daptomycin is a novel
cyclic lipopeptide fermentation product of Streptomyces
roseosporus (Figure 43–8). It was discovered decadesago but has only
recently been developed as the need for drugs
Its spec-trum of activity is similar
to that of vancomycin except that it is more rapidly bactericidal in vitro and
may be active against vancomycin-resistant strains of enterococci and S aureus. The precise mechanism of
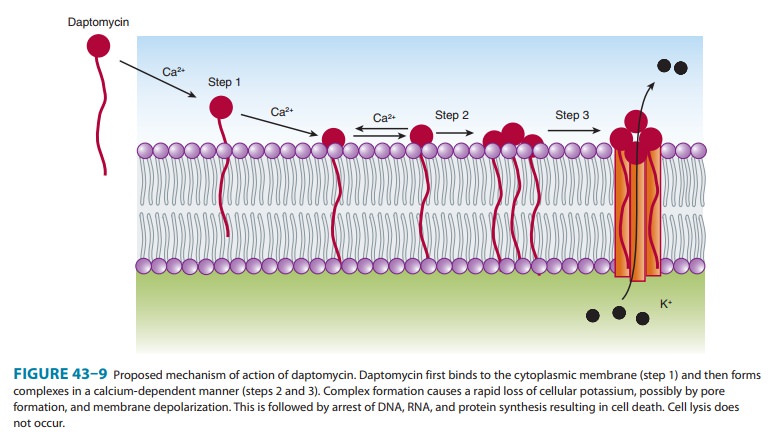
action is not fully understood, but it is known to bind to the cell membrane
via calcium-dependent inser-tion of its lipid tail. This results in depolarization
of the cell mem-brane with potassium efflux and rapid cell death (Figure 43–9).
Daptomycin is cleared renally. The approved doses are 4 mg/kg/ dose for
treatment of skin and soft tissue infections and 6 mg/kg/ dose for treatment of
bacteremia and endocarditis once daily in patients with normal renal function
and every other day in patients with creatinine clearance of less than 30
mL/min. Forserious infections, many experts recommend using doses of
dapto-mycin higher than 6 mg/kg/dose. In clinical trials powered for
noninferiority, daptomycin was equivalent in efficacy to vancomy-cin. It can
cause myopathy, and creatine phosphokinase levels should be monitored weekly.
Pulmonary surfactant antagonizes daptomycin, and it should not be used to treat
pneumonia. Daptomycin can also cause an allergic pneumonitis in patients
receiving prolonged therapy (> 2 weeks). Treatment failures have been reported in association
with an increase in daptomycin MIC for clinical isolates obtained during
therapy, but the relation between the increase in MIC and treatment failure is
unclear at this point. Daptomycin is an effective alternative to vancomycin,
and its ultimate role continues to unfold.
FOSFOMYCIN
Fosfomycin trometamol,
a stable salt of fosfomycin (phosphono-mycin), inhibits a very early stage of
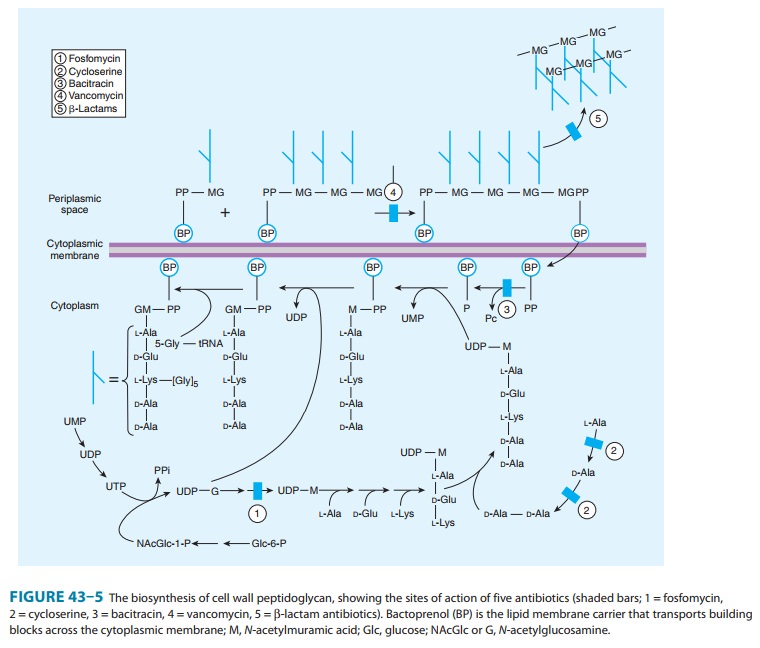
bacterial cell wall synthesis (Figure 43–5). An analog of phosphoenolpyruvate,
it is structurally unrelated to any other antimicrobial agent. It inhibits the
cytoplas-mic enzyme enolpyruvate transferase by covalently binding to the
cysteine residue of the active site and blocking the addition of
phosphoenolpyruvate to UDP-N-acetylglucosamine.
This reaction is the first step in the formation of UDP-N-acetylmuramic acid, the precursor of N-acetylmuramic acid, which is found only in bacterial cell walls.
The drug is transported into the bacterial cell by glycerophosphate or glucose
6-phosphate transport systems. Resistance is due to inadequate transport of
drug into the cell.
Fosfomycin is active
against both gram-positive and gram-negative organisms at concentrations ≥ 125 mcg/mL.
Susceptibility tests should be performed in growth medium supplemented with
glucose 6-phosphate to minimize false-positive indications of resistance. In
vitro synergism occurs when fosfomycin is combined with β-lactam antibiotics,
aminoglycosides, or fluoroquinolones.
Fosfomycin trometamol
is available in both oral and parenteral formulations, although only the oral
preparation is approved for use in the USA. Oral bioavailability is
approximately 40%. Peak serum concentrations are 10 mcg/mL and 30 mcg/mL
following a 2-g or 4-g oral dose, respectively. The half-life is approximately
4 hours. The active drug is excreted by the kidney, with urinary concentrations
exceeding MICs for most urinary tract pathogens.
Fosfomycin is approved
for use as a single 3-g dose for treat-ment of uncomplicated lower urinary
tract infections in women. The drug appears to be safe for use in pregnancy.
BACITRACIN
Bacitracin is a cyclic
peptide mixture first obtained from the Tracy strain of Bacillus subtilis in 1943. It is active against gram-positive
microorganisms. Bacitracin inhibits cell wall formation by inter-fering with
dephosphorylation in cycling of the lipid carrier that transfers peptidoglycan
subunits to the growing cell wall (Figure 43–5). There is no cross-resistance
between bacitracin and other antimicrobial drugs.
Bacitracin is highly
nephrotoxic when administered systemi-cally and is only used topically.
Bacitracin is poorly absorbed. Topical application results in local
antibacterial activity without systemic toxicity. Bacitracin, 500 units/g in an
ointment base (often combined with polymyxin or neomycin), is indicated for the
suppression of mixed bacterial flora in surface lesions of the skin, in wounds,
or on mucous membranes. Solutions of bacitracin containing 100–200 units/mL in
saline can be used for irrigation of joints, wounds, or the pleural cavity.
CYCLOSERINE
Cycloserine is an
antibiotic produced by Streptomyces
orchidaceous. It is water soluble and very unstable at acid pH. Cycloserine
inhib-its many gram-positive and gram-negative organisms, but it is used almost
exclusively to treat tuberculosis caused by strains of Mycobacterium tuberculosis resistant to first-line agents.
Cycloserine isa structural analog of D-alanine
and inhibits the incorporation of D-alanine
into peptidoglycan pentapeptide by inhibiting alanine race-mase, which converts
L-alanine to D-alanine, and D-alanyl- D-alanine ligase
(Figure 43–5). After ingestion of 0.25 g of cycloserine blood levels reach
20–30 mcg/mL—sufficient to inhibit many strains of mycobacteria and
gram-negative bacteria. The drug is widely distributed in tissues. Most of the
drug is excreted in active form into the urine. The dosage for treating
tuberculosis is 0.5 to 1 g/d in two or three divided doses.
Cycloserine causes
serious dose-related central nervous system toxicity with headaches, tremors,
acute psychosis, and convul-sions. If oral dosages are maintained below 0.75
g/d, such effects can usually be avoided.
Related Topics