Chapter: Basic & Clinical Pharmacology : Beta-Lactam & OtherCell Wall- & Membrane-Active Antibiotics
Glycopeptide Antibiotics
GLYCOPEPTIDE ANTIBIOTICS
VANCOMYCIN
Vancomycin
is an antibiotic produced by Streptococcus
orientalis and Amycolatopsis
orientalis. With the exception of Flavobacterium,
it is active only against gram-positive bacteria. Vancomycin is a glycopeptide
of molecular weight 1500. It is water soluble and quite stable.
Mechanisms of Action & Basis of Resistance
Vancomycin inhibits
cell wall synthesis by binding firmly to the D-Ala-D-Ala terminus of nascent peptidoglycan pentapeptide(Figure
43–5). This inhibits the transglycosylase, preventing fur-ther elongation of
peptidoglycan and cross-linking. The peptido-glycan is thus weakened, and the
cell becomes susceptible to lysis. The cell membrane is also damaged, which
contributes to the antibacterial effect.
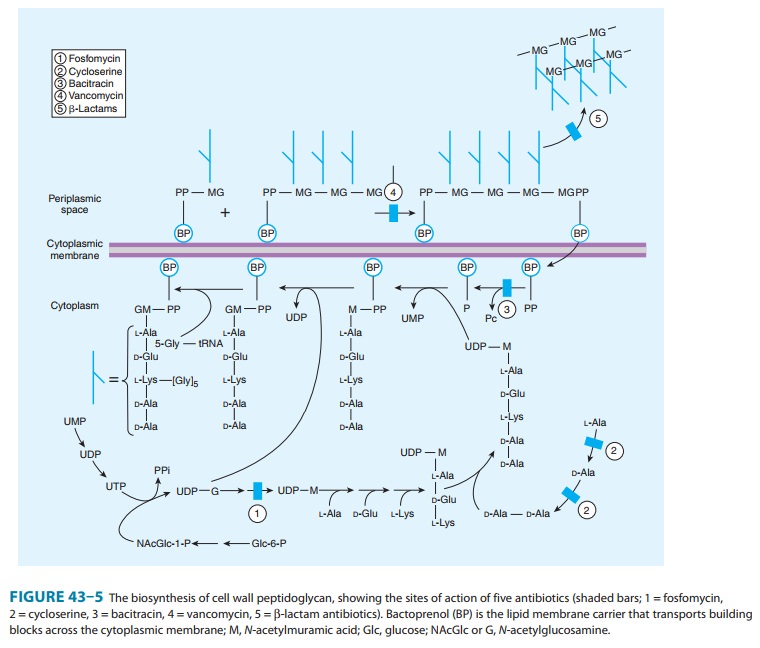
Resistance to
vancomycin in enterococci is due to modification of the D-Ala-D-Ala binding site of
the peptidoglycan building block in which the terminal D-Ala is replaced by D-lactate.
This results in the loss of a critical hydrogen bond that facilitates
high-affinity binding of vancomycin to its target and loss of activity. This
mechanism is also present in vancomycin-resistant S aureus strains (MIC ≥ 16 mcg/mL), which have acquired the
enterococ-cal resistance determinants. The underlying mechanism for reduced
vancomycin susceptibility in vancomycin-intermediate strains (MICs = 4–8 mcg/mL) of S aureus is not fully known. However
these strains have altered cell wall metabolism that results in a
thickened cell wall with increased numbers of D-Ala-D-Ala residues, which serve
as dead-end binding sites for vanco-mycin. Vancomycin is sequestered within the
cell wall by these false targets and may be unable to reach its site of action.
Antibacterial Activity
Vancomycin
is bactericidal for gram-positive bacteria in concentra-tions of 0.5–10 mcg/mL.
Most pathogenic staphylococci, includ-ing those producing β lactamase
and those resistant to nafcillin and methicillin, are killed by 2 mcg/mL or
less. Vancomycin kills staphylococci relatively slowly and only if cells are
actively dividing; the rate is less than that of the penicillins both in vitro
and in vivo. Vancomycin is synergistic in vitro with gentamicin and streptomy-cin
against Enterococcus faecium and Enterococcus faecalis strains that do
not exhibit high levels of aminoglycoside resistance.
Pharmacokinetics
Vancomycin
is poorly absorbed from the intestinal tract and is administered orally only
for the treatment of antibiotic-associated colitis caused by C difficile. Parenteral doses must be
administered intravenously. A 1-hour intravenous infusion of 1 g produces blood
levels of 15–30 mcg/mL for 1–2 hours. The drug is widely distributed in the
body. Cerebrospinal fluid levels 7–30% of simultaneous serum concentrations are
achieved if there is menin-geal inflammation. Ninety percent of the drug is
excreted by glomerular filtration. In the presence of renal insufficiency,
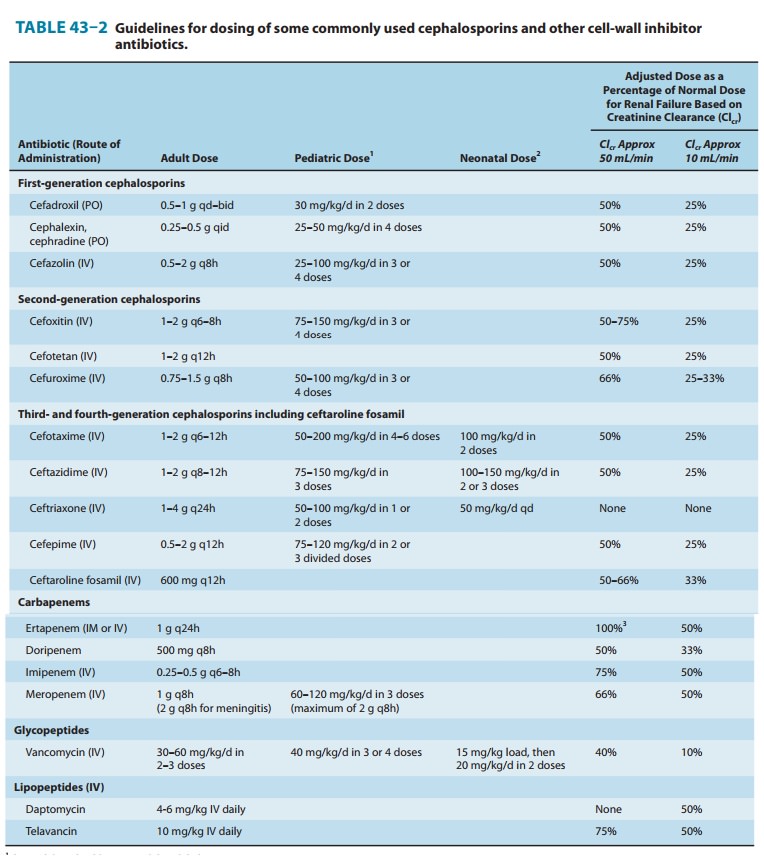
strik-ing accumulation may occur (Table 43–2). In functionally anephric
patients, the half-life of vancomycin is 6–10 days. A significant amount
(roughly 50%) of vancomycin is removed during a standard hemodialysis run when
a modern, high-flux membrane is used.
Clinical Uses
Important indications
for parenteral vancomycin are bloodstream infections and endocarditis caused by
methicillin-resistant staphy-lococci. However, vancomycin is not as effective
as an antistaphy-lococcal penicillin for treatment of serious infections such
as endocarditis caused by methicillin-susceptible strains. Vancomycin in
combination with gentamicin is an alternative regimen for treatment of
enterococcal endocarditis in a patient with serious penicillin allergy.
Vancomycin (in combination with cefotaxime, ceftriaxone, or rifampin) is also
recommended for treatment of meningitis suspected or known to be caused by a
penicillin-resis-tant strain of pneumococcus (ie, penicillin MIC > 1 mcg/mL). The
recommended dosage in a patient with normal renal function is 30–60 mg/kg/d in
two or three divided doses. The traditional dosing regimen in adults with
normal renal function is 1 g every 12 hours (∼ 30 mg/kg/d); however, this dose will not
typically achieve the trough concentrations (15–20 mcg/mL) recom-mended for
serious infections. For serious infections , a starting dose of 45–60 mg/kg/d
should be given with titration of the dose to achieve trough levels of 15–20
mcg/mL. The dosage in children is 40 mg/kg/d in three or four divided doses.
Clearance of vancomycin is directly proportional to creatinine clearance, and
the dosage is reduced accordingly in patients with renal insuffi-ciency. For
functionally anephric adult patients, a 1-g dose admin-istered once a week may
be sufficient. For patients receiving hemodialysis, a common dosing regimen is
a 1-g loading dose fol-lowed by 500 mg after each dialysis session. Patients
receiving a prolonged course of therapy should have serum concentrations
checked. Recommended trough concentrations are 10–15 mcg/mL for mild to
moderate infections such as cellulitis and 15–20 mcg/mL for more serious
infections such as endocarditis, meningitis, and necrotizing pneumonia.
Oral vancomycin,
0.125–0.25 g every 6 hours, is used to treat antibiotic-associated colitis
caused by C difficile. Because of the
emergence of vancomycin-resistant enterococci and the potential selective
pressure of oral vancomycin for these resistant organisms, metronidazole had
been preferred as initial therapy over the last two decades. However, receipt
of oral vancomycin does not appear to be a significant risk factor for
acquisition of vancomycin-resistant enterococci. Additionally, recent clinical
data suggest that vancomycin is associated with a better clinical response than
metronidazole for more severe cases of C
difficile colitis. Therefore, oral vancomycin may be used as a first line
treatment for severe cases or for cases that fail to respond to metronidazole.
Adverse Reactions
Adverse
reactions are encountered in about 10% of cases. Most reactions are minor.
Vancomycin is irritating to tissue, resulting in phlebitis at the site of
injection. Chills and fever may occur. Ototoxicity is rare and nephrotoxicity
uncommon with current preparations. However, administration with another
ototoxic or nephrotoxic drug, such as an aminoglycoside, increases the risk of
these toxicities. Ototoxicity can be minimized by maintaining peak serum
concentrations below 60 mcg/mL. Among the more common reactions is the
so-called “red man” or “red neck” syndrome. This infusion-related flushing is
caused by release of histamine. It can be largely prevented by prolonging the
infusion period to 1–2 hours or pretreatment with an antihistamine such as
diphenhydramine.
TEICOPLANIN
Teicoplanin
is a glycopeptide antibiotic that is very similar to vancomycin in mechanism of
action and antibacterial spectrum. Unlike vancomycin, it can be given
intramuscularly as well as intravenously. Teicoplanin has a long half-life
(45–70 hours), per-mitting once-daily dosing. This drug is available in Europe
but has not been approved for use in the United States.
TELAVANCIN
Telavancin
is a semisynthetic lipoglycopeptide derived from vancomycin. Telavancin is
active versus gram-positive bacteria, including strains with reduced
susceptibility to vancomycin. Telavancin has two mechanisms of action. Like
vancomycin, tela-vancin inhibits cell wall synthesis by binding to the D-Ala-D-Ala
terminus of peptidoglycan in the growing cell wall. In addition, it disrupts
the bacterial cell membrane potential and increases mem-brane permeability. The
half-life of telavancin is approximately 8 hours, which supports once-daily
intravenous dosing. Telavancin is approved for treatment of complicated skin
and soft tissue infec-tions at a dose of 10 mg/kg IV daily. Unlike vancomycin
therapy, monitoring of serum telavancin levels is not required. Telavancin is
potentially teratogenic, so administration to pregnant women must be avoided.
DALBAVANCIN
Dalbavancin is a
semisynthetic lipoglycopeptide derived from teicoplanin. Dalbavancin shares the
same mechanism of action as vancomycin and teicoplanin but has improved
activity against many gram-positive bacteria including methicillin-resistant
and vancomycin-intermediate S aureus.
It is not active against most strains of vancomycin-resistant enterococci.
Dalbavancin has an extremely long half-life of 6–11 days, which allows for
once-weekly intravenous administration. Development of dalbavancin has been put
on hold pending additional clinical trials.
Related Topics