Chapter: Medical Physiology: Renal Regulation of Potassium, Calcium, Phosphate, and Magnesium; Integration of Renal Mechanisms for Control of Blood Volume and Extracellular Fluid Volume
Nervous and Hormonal Factors Increase the Effectiveness of Renal-Body Fluid Feedback Control
Nervous and Hormonal Factors Increase the Effectiveness of Renal-Body Fluid Feedback Control
We discuss the nervous and hormonal factors that influence GFR and tubular reabsorption and, therefore, renal excretion of salt and water. These nervous and hormonal mechanisms usually act in concert with the pressure natriuresis and pressure diuresis mechanisms, making them more effective in minimizing the changes in blood volume, extracellular fluid volume, and arterial pressure that occur in response to day-to-day challenges. However, abnor-malities of kidney function or of the various nervous and hormonal factors that influence the kidneys can lead to serious changes in blood pressure and body fluid volumes, as discussed later.
Sympathetic Nervous System Control of Renal Excretion: Arterial Baroreceptor and Low-Pressure Stretch Receptor Reflexes
Because the kidneys receive extensive sympathetic innervation, changes in sympathetic activity can alter renal sodium and water excretion as well as regulation of extracellular fluid volume under some conditions. For example, when blood volume is reduced by hem-orrhage, the pressures in the pulmonary blood vessels and other low-pressure regions of the thorax decrease, causing reflex activation of the sympathetic nervous system. This in turn increases renal sympathetic nerve activity, which has several effects to decrease sodium and water excretion: (1) constriction of the renal arte-rioles, with resultant decreased GFR; (2) increased tubular reabsorption of salt and water; and (3) stimu-lation of renin release and increased angiotensin II and aldosterone formation, both of which further increase tubular reabsorption. And if the reduction in blood volume is great enough to lower systemic arte-rial pressure, further activation of the sympathetic nervous system occurs because of decreased stretch of the arterial baroreceptors located in the carotid sinus and aortic arch. All these reflexes together play an important role in the rapid restitution of blood volume that occurs in acute conditions such as hemorrhage. Also, reflex inhibition of renal sympathetic activity may contribute to the rapid elimination of excess fluid in the circulation that occurs after eating a meal that contains large amounts of salt and water.
Role of Angiotensin II In Controlling Renal Excretion
One of the body’s most powerful controllers of sodium excretion is angiotensin II. Changes in sodium and fluid intake are associated with reciprocal changes in angiotensin II formation, and this in turn contributes greatly to the maintenance of body sodium and fluid balances. That is, when sodium intake is elevated above normal, renin secretion is decreased, causing decreased angiotensin II formation. Because angiotensin II has several important effects in increas-ing tubular reabsorption of sodium, a reduced level of angiotensin II decreases tubular reabsorption of sodium and water, thus increasing the kidneys’ excretion of sodium and water. The net result is to minimize the rise in extracellular fluid volume and arterial pressure that would other-wise occur when sodium intake increases.
Conversely, when sodium intake is reduced below normal, increased levels of angiotensin II cause sodium and water retention and oppose reductions in arterial blood pressure that would otherwise occur. Thus, changes in activity of the renin-angiotensin system act as a powerful amplifier of the pressure natriuresis mechanism for maintaining stable blood pressures and body fluid volumes.
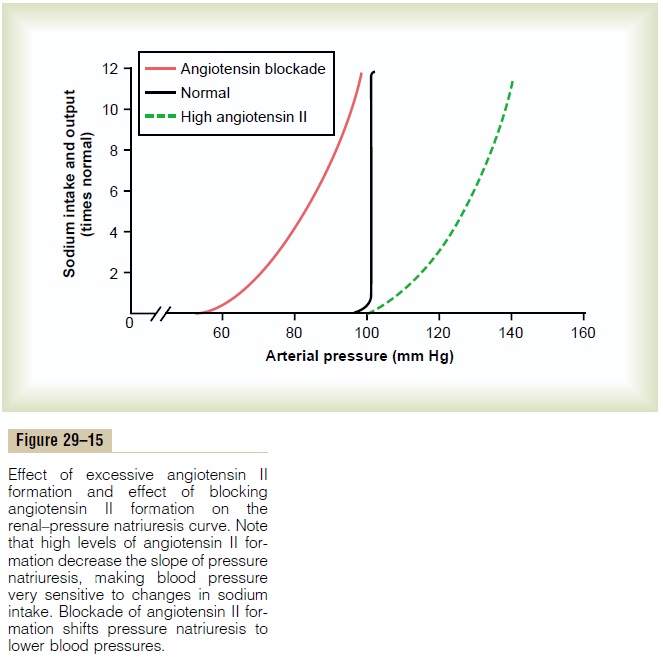
Importance of Angiotensin II in Increasing Effectiveness of Pressure Natriuresis. The importance of angiotensin IIin making the pressure natriuresis mechanism more effective is shown in Figure 29–15. Note that when the angiotensin control of natriuresis is fully functional, the pressure natriuresis curve is steep (normal curve), indicating that only minor changes in blood pressure are needed to increase sodium excretion when sodium intake is raised.
In contrast, when angiotensin levels cannot be decreased in response to increased sodium intake (high angiotensin II curve), as occurs in some hyper-tensive patients who have impaired ability to decrease renin secretion, the pressure natriuresis curve is not nearly as steep. Therefore, when sodium intake is raised, much greater increases in arterial pressure are needed to increase sodium excretion and maintain sodium balance. For example, in most people, a 10-fold increase in sodium intake causes an increase of only a few millimeters of mercury in arterial pressure, whereas in subjects who cannot suppress angiotensin
II formation appropriately in response to excess sodium, the same rise in sodium intake causes blood pressure to rise as much as 50 mm Hg. Thus, the inabil-ity to suppress angiotensin II formation when there is excess sodium reduces the slope of pressure natriure-sis and makes arterial pressure very salt sensitive.
The use of drugs to block the effects of angiotensin II has proved to be important clinically for improv-ing the kidneys’ ability to excrete salt and water. When angiotensin II formation is blocked with an angiotensin-converting enzyme inhibitor (see Figure 29–15) or an angiotensin II receptor antagonist, the renal–pressure natriuresis curve is shifted to lower pressures; this indicates an enhanced ability of the kidneys to excrete sodium because normal levels of sodium excretion can now be maintained at reduced arterial pressures. This shift of pressure natriuresis provides the basis for the chronic blood pressure–lowering effects in hypertensive patients of the angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists.
Excessive Angiotensin II Does Not Cause Large Increases in Extracellular Fluid Volume Because Increased Arterial Pressure Counterbalances Angiotensin-Mediated Sodium Retention.
Although angiotensin II is one of the most powerful sodium- and water-retaining hormones in the body, neither a decrease nor an increase in circulating angiotensin II has a large effect on extracellular fluid volume or blood volume. The reason for this is that with large increases in angiotensin II levels, as occurs with a renin-secreting tumor of the kidney, the high angiotensin II levels initially cause sodium and water retention by the kidneys and a small increase in extracellular fluid volume. This also initi-ates a rise in arterial pressure that quickly increases kidney output of sodium and water, thereby overcom-ing the sodium- and water-retaining effects of the angiotensin II and re-establishing a balance between intake and output of sodium at a higher blood pres-sure. Conversely, after blockade of angiotensin II for-mation, as occurs when an angiotensin-converting enzyme inhibitor is administered, there is initial loss of sodium and water, but the fall in blood pressure offsets this effect, and sodium excretion is once again restored to normal.
Role of Aldosterone in Controlling Renal Excretion
Aldosterone increases sodium reabsorption, especially in the cortical collecting tubules. The increased sodium reabsorption is also associated with increased water reabsorption and potassium secretion. Therefore, the net effect of aldosterone is to make the kidneys retain sodium and water but to increase potassium excretion in the urine.
The function of aldosterone in regulating sodium balance is closely related to that described for angiotensin II.That is, with reduction in sodium intake, the increased angiotensin II levels that occur stimulate aldosterone secretion, which in turn contributes to the reduction in urinary sodium excretion and, therefore, to the maintenance of sodium balance. Conversely, with high sodium intake, suppression of aldosterone formation decreases tubular reabsorption, allowing the kidneys to excrete larger amounts of sodium. Thus, changes in aldosterone formation also aid the pressure natriuresis mechanism in maintaining sodium balance during variations in salt intake.
During Chronic Oversecretion of Aldosterone, Kidneys “Escape” from Sodium Retention as Arterial Pressure Rises.
Although aldosterone has powerful effects on sodium reabsorption, when there is excessive infusion of aldosterone or excessive formation of aldosterone, as occurs in patients with tumors of the adrenal gland (Conn’s syndrome), the increased sodium reabsorp-tion and decreased sodium excretion by the kidneys are transient. After 1 to 3 days of sodium and water retention, the extracellular fluid volume rises by about 10 to 15 per cent and there is a simultaneous increase in arterial blood pressure. When the arterial pressure rises sufficiently, the kidneys “escape” from the sodium and water retention and thereafter excrete amounts of sodium equal to the daily intake, despite continued presence of high levels of aldosterone. The primary reason for the escape is the pressure natriuresis and diuresis that occur when the arterial pressure rises.
In patients with adrenal insufficiency who do not secrete enough aldosterone (Addison’s disease), there is increased excretion of sodium and water, reduction in extracellular fluid volume, and a tendency toward low blood pressure. In the complete absence of aldos-terone, the volume depletion may be severe unless the person is allowed to eat large amounts of salt and drink large amounts of water to balance the increased urine output of salt and water.
Role of ADH in Controlling Renal Water Excretion
As discussed, ADH plays an important role in allowing the kidneys to form a small volume of concentrated urine while excreting normal amounts of salt. This effect is especially important during water deprivation, which strongly elevates plasma levels of ADH that in turn increase water reabsorption by the kidneys and help to minimize the decreases in extra-cellular fluid volume and arterial pressure that would otherwise occur. Water deprivation for 24 to 48 hours normally causes only a small decrease in extracellular fluid volume and arterial pressure. However, if the effects of ADH are blocked with a drug that antago-nizes the action of ADH to promote water reabsorp-tion in the distal and collecting tubules, the same period of water deprivation causes a substantial fall in both extracellular fluid volume and arterial pressure.
Conversely, when there is excess extracellular volume, decreased ADH levels reduce reabsorption of water bythe kidneys, thus helping to rid the body of the excess volume.
Excess ADH Secretion Usually Causes Only Small Increases in Extracellular Fluid Volume but Large Decreases in Sodium Con- centration. Although ADH is important in regulatingextracellular fluid volume, excessive levels of ADH seldom cause large increases in arterial pressure or extracellular fluid volume. Infusion of large amounts of ADH into animals initially causes renal retention of water and a 10 to 15 per cent increase in extracellular fluid volume. As the arterial pressure rises in response to this increased volume, much of the excess volume is excreted because of the pressure diuresis mecha-nism. After several days of ADH infusion, the blood volume and extracellular fluid volume are elevated no more than 5 to 10 per cent, and the arterial pressure is also elevated by less than 10 mm Hg. The same is true for patients withinappropriate ADH syndrome, in which ADH levels may be elevated severalfold.
Thus, high levels of ADH do not cause major increases of either body fluid volume or arterial pres-sure, although high ADH levels can cause severe reduc-tions in extracellular sodium ion concentration. Thereason for this is that increased water reabsorption by the kidneys dilutes the extracellular sodium, and at the same time, the small increase in blood pressure that does occur causes loss of sodium from the extracellu-lar fluid in the urine through pressure natriuresis.
In patients who have lost their ability to secrete ADH because of destruction of the supraoptic nuclei, the urine volume may become 5 to 10 times normal. This is almost always compensated for by ingestion of enough water to maintain fluid balance. If free access to water is prevented, the inability to secrete ADH may lead to marked reductions in blood volume and arterial pressure.
Role of Atrial Natriuretic Peptide in Controlling Renal Excretion
Thus far, we have discussed mainly the role of sodium-and water-retaining hormones in controlling extracel-lular fluid volume. However, several different natri-uretic hormones may also contribute to volume regulation. One of the most important of the natri-uretic hormones is a peptide referred to as atrial natri-uretic peptide (ANP), released by the cardiac atrialmuscle fibers. The stimulus for release of this peptide appears to be overstretch of the atria, which can result from excess blood volume. Once released by the cardiac atria, ANP enters the circulation and acts on the kidneys to cause small increases in GFR and decreases in sodium reabsorption by the collecting ducts. These combined actions of ANP lead to increased excretion of salt and water, which helps to compensate for the excess blood volume.
Changes in ANP levels probably help to minimize changes in blood volume during various disturbances, such as increased salt and water intake. However, excessive production of ANP or even complete lack of ANP does not cause major changes in blood volume because these effects can easily be overcome by small changes in blood pressure, acting through pressure natriuresis. For example, infusions of large amounts of ANP initially raise urine output of salt and water and cause slight decreases in blood volume. In less than 24 hours, this effect is overcome by a slight decrease in blood pressure that returns urine output toward normal, despite continued excess of ANP.
Related Topics