Chapter: Basic & Clinical Pharmacology : Adrenoceptor Agonists & Sympathomimetic Drugs
Molecular Pharmacology Underlying the Actions of Sympathomimetic Drugs
MOLECULAR
PHARMACOLOGY UNDERLYING THE ACTIONS OF SYMPATHOMIMETIC DRUGS
The
effects of catecholamines are mediated by cell-surface receptors.
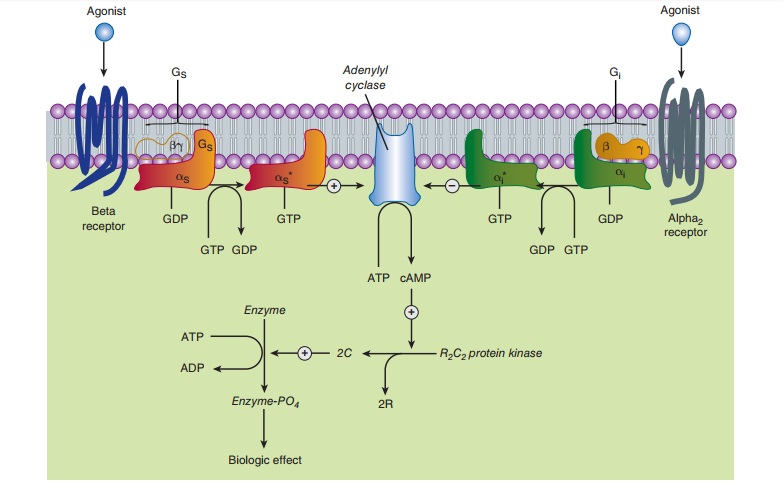
Adrenoceptors are typical G protein-coupled receptors (GPCRs;). The receptor
protein has an extracellular N-terminus, traverses the membrane seven times
(transmembrane domains) forming three extracellular and three intracellular
loops, and has an intracellular C-terminus (Figure 9–1). G proteincoupled
receptors are coupled by G proteins to the various effec-tor proteins whose
activities are regulated by those receptors. EachG protein is a heterotrimer
consisting of α,
β,
and γ
subunits. G proteins are classified on the basis of their distinctive α subunits. G proteins
of particular importance for adrenoceptor function include Gs, the
stimulatory G protein of adenylyl cyclase; Gi and Go, the
inhibitory G proteins of adenylyl cyclase; and Gq and G11,
the G proteins coupling α receptors to phospholipase C. The activation
of G protein-coupled receptors by catecholamines pro-motes the dissociation of
guanosine diphosphate (GDP) from thesubunit of the appropriate G protein.
Guanosine triphosphate (GTP) then binds to this G protein, and the α subunit dissociates
from the β-γ unit. The activated
GTP-bound α
subunit then regulates the activity of its effector. Effectors of
adrenoceptor-activated α subunits include adenylyl cyclase, cGMP
phosphodi-esterase, phospholipase C, and ion channels. The α subunit is
inactivated by hydrolysis of the bound GTP to GDP and phos-phate, and the
subsequent reassociation of the α subunit with the β-γ subunit. The β-γ subunits have additional independent
effects,acting on a variety of effectors such as ion channels and enzymes.
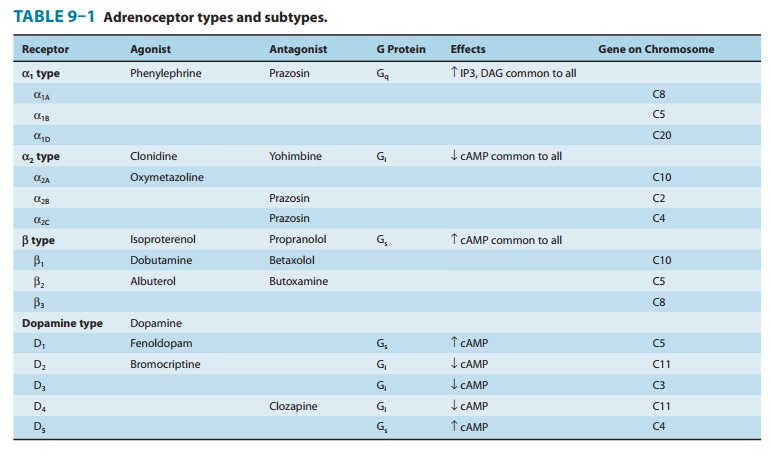
Adrenoreceptors were initially characterized pharmacologically, with α receptors having the comparative potencies epinephrine ≥ norepinephrine >> isoproterenol, and β receptors having the com-parative potencies isoproterenol > epinephrine ≥ norepinephrine. The development of selective antagonists revealed the presence of subtypes of these receptors, which were finally characterized by molecular cloning. We now know that unique genes encode the receptor subtypes listed in Table 9–1.
Likewise,
the endogenous catecholamine dopamine produces a variety of biologic effects
that are mediated by interactions with specific dopamine receptors (Table 9–1).
These receptors are dis-tinct from α and β receptors and are particularly important in
the brain and in the splanchnic and
renal vasculature. Molecular cloning has identified several distinct genes
encoding five receptor subtypes, two D1-like receptors (D1
and D5) and three D2-like (D2, D3,
and D4). Further complexity occurs because of the presence of
introns within the coding region of the D2-like receptor genes,
which allows for alternative splicing of the exons in this major subtype. There
is extensive polymorphic variation in the D4 human receptor gene.
These subtypes may have importance for understanding the efficacy and adverse
effects of novel antipsychotic drugs .
Receptor Types
A. Alpha Receptors
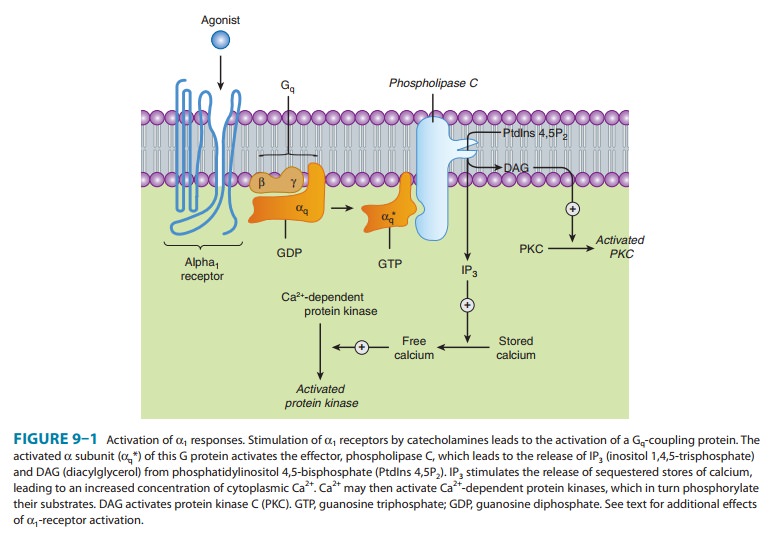
Alpha1
receptors are coupled via G proteins in the Gq family to
phospholipase C. This enzyme hydrolyzes polyphosphoinositides, leading to the
formation of inositol
1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (Table 9–1, Figure 9–1). IP3promotes
therelease of sequestered Ca2+ from intracellular
stores, which increases the cytoplasmic concentration of free Ca2+ and the activation of
various calcium-dependent protein kinases. Activation of these receptors may
also increase influx of calcium across the cell’s plasma
membrane.
IP3 is sequentially dephosphorylated, which ultimately leads to the
formation of free inositol. DAG activates protein kinase C, which modulates
activity of many signaling pathways. In addi-tion, α1 receptors activate signal transduction
pathways that were originally described for peptide growth factor receptors
that activate tyrosine kinases. For example, α1 receptors have been found to acti-vate
mitogen-activated kinases (MAP kinases) and polyphospho-inositol-3-kinase
(PI-3-kinase). These pathways may have importance for the α1-receptor–mediated
stimulation of cell growth and proliferation through the regulation of gene
expression.
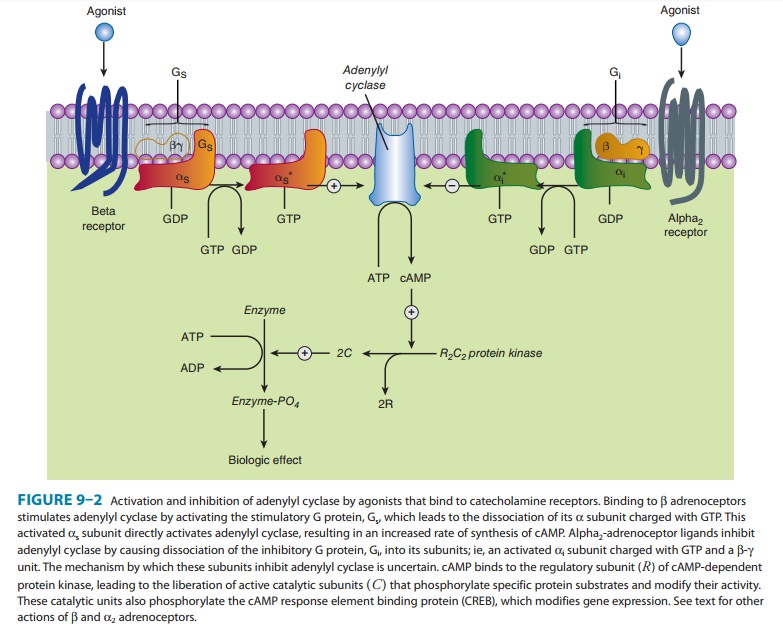
Alpha2
receptors inhibit adenylyl cyclase activity and cause intracellular cyclic
adenosine monophosphate (cAMP) levels to decrease. Alpha2-receptor–mediated
inhibition of adenylyl cyclase activity is transduced by the inhibitory
regulatory protein, Gi (Figure 9–2). It is likely that not only α, but also the β-γ subunits of Gi
contribute to inhibition of adenylyl cyclase. Alpha2 receptors use
other signaling pathways, including regulation of ion channel activities and
the activities of important enzymes involved in sig-nal transduction. Indeed,
some of the effects of α2 adrenoceptors are independent of their
ability to inhibit adenylyl cyclase; for example, α2-receptor agonists cause platelet aggregation
and a decrease in platelet cAMP levels, but it is not clear whether
aggre-gation is the result of the decrease in cAMP or other mechanisms
involving Gi-regulated effectors.
B. Beta Receptors
Activation
of all three receptor subtypes (β1, β2, and β3) results in stimulation of adenylyl cyclase
and increased conversion ofadenosine triphosphate (ATP) to cAMP (Table 9–1,
Figure 9–2). Activation of the cyclase enzyme is mediated by the stimulatory
coupling protein Gs. Cyclic AMP is the major second messenger of β-receptor activation.
For example, in the liver of many species, β-receptor–activated cAMP synthesis leads to a
cascade of eventsculminating in the activation of glycogen phosphorylase. In
the heart, β-receptor–activated
cAMP synthesis increases the influx of calcium across the cell membrane and its
sequestration inside the cell. Beta-receptor activation also promotes the
relaxation of smooth muscle. Although the mechanism of the smooth muscle effect
is uncertain, it may involve the phosphorylation of myosin light-chain kinase
to an inactive form (see Figure 12–1). Beta adrenoceptors may activate voltage-sensitive
calcium channels in the heart via Gs-mediated enhancement
independently of changes in cAMP concentration. Under certain circumstances, β2 recep-tors may couple
to Gq proteins. These receptors have been dem-onstrated to activate
additional kinases, such as MAP kinases, byforming multi-subunit complexes
within cells, which contain multiple signaling molecules.
The
β3 adrenoreceptor is a lower affinity receptor compared with β1 and β2 receptors but is more
resistant to desensitization. It is found in several tissues, but its
physiologic or pathologic role in humans is not clear. Selective agonists and
antagonists have been developed but are not clinically available.
C. Dopamine Receptors
The D1 receptor is typically associated with the stimulation of adenylyl cyclase (Table 9–1); for example, D1-receptor–induced smooth muscle relaxation is presumably due to cAMP accumula-tion in the smooth muscle of those vascular beds in which dop-amine is a vasodilator. D2 receptors have been found to inhibit adenylyl cyclase activity, open potassium channels, and decrease calcium influx.
Receptor Selectivity
Many
clinically available adrenergic agonists have selectivity for the major (α1 and α2 versus β) adrenoreceptor
types, but not for the subtypes of these major groups. Examples of clinically
useful sympathomimetic agonists that are relatively selective for α1-, α2-, and β-adrenoceptor
subgroups are compared with some nonselec-tive agents in Table 9–2. Selectivity
means that a drug may prefer-entially bind to one subgroup of receptors at
concentrations too low to interact extensively with another subgroup. However,
selec-tivity is not usually absolute (nearly absolute selectivity has been
termed “specificity”), and at higher concentrations, a drug may also interact
with related classes of receptors. The effects of a given drug may depend not
only on its selectivity to adrenoreceptor types, but also to the relative
expression of receptor subtypes in a given tissue. (see Box: Receptor
Selectivity and Physiologic Functions of Adrenoceptor Subtypes).
Receptor Regulation
Responses
mediated by adrenoceptors are not fixed and static. The number and function of
adrenoceptors on the cell surface and their responses may be regulated by
catecholamines themselves, other hormones and drugs, age, and a number of
disease states . These changes may modify the magnitude of a tissue’s
physiologic response to catecholamines and can be important clinically during
the course of treatment. One of the best-studied examples of receptor
regulation is the desensitization of
adreno-ceptors that may occur after exposure to catecholamines and other
sympathomimetic drugs. After a cell or tissue has been exposed for a period of
time to an agonist, that tissue often becomes less responsive to further
stimulation by that agent (see Figure 2–12).
Receptor Selectivity and Physiologic Functions of Adrenoceptor Subtypes: Lessons from Knockout Mice
Since pharmacologic tools used to evaluate the
function of adrenoceptor subtypes have some limitations, a number of knockout
mice have been developed with one or more adre-noceptor genes subjected to loss
of function mutations, as described (see Box: Pharmacology & Genetics).
These models have their own complexities, and extrapola-tions from mice to
humans may be uncertain. Nonetheless, these studies have yielded some novel
insights. For example, α-adrenoceptor subtypes play an important role in
cardiac responses, the α2A-adrenoceptor subtype is critical in
trans-ducing the effects of α2 agonists on blood pressure control,
and β1 receptors play a predominant role in directly increas-ing
heart rate in the mouse heart.
Other terms
such as tolerance, refractoriness, and tachyphylaxis have also been used to
denote desensitization. This process has potential clinical significance
because it may limit the therapeutic response to sympathomimetic agents.
Many
mechanisms have been found to contribute to desensiti-zation. Some mechanisms
function relatively slowly, over the course of hours or days, and these
typically involve transcriptional or translational changes in the receptor
protein level, or its migra-tion to the cell surface. Other mechanisms of
desensitization occur quickly, within minutes. Rapid modulation of receptor
function in desensitized cells may involve critical covalent modification of
the receptor, especially by phosphorylation on specific amino acid residues,
association of these receptors with other proteins, or changes in their
subcellular location.
There
are two major categories of desensitization of responses mediated by G
protein-coupled receptors. Homologous
desensi-tization refers to loss of responsiveness exclusively of the receptors
that have been exposed to repeated or sustained activation by an agonist. Heterologous desensitization refers to
the process by which desensitization of one receptor by its agonists also
results in desensitization of another receptor that has not been directly
acti-vated by the agonist in question.
A
major mechanism of desensitization that occurs rapidly involves phosphorylation
of receptors by members of the G
protein-coupled receptor kinase (GRK) family, of which there are
sevenmembers. Specific adrenoceptors become substrates for these kinases only
when they are bound to an agonist. This mechanism is an example of homologous
desensitization because it specifically involves only agonist-occupied
receptors.
Phosphorylation
of these receptors enhances their affinity for arrestins, a family of four widely expressed proteins. Upon binding
of arrestin, the capacity of the receptor to activate G proteins is blunted,
presumably as a result of steric hindrance(see Figure 2–12). Arrestin then
interacts with clathrin and clathrin adaptor AP2, leading to endocytosis of the
receptor. In addition to blunting responses requiring the presence of the
receptor on the cell surface, these regulatory processes may also contribute to
novel mechanisms of receptor signaling via intra-cellular pathways.
Receptor
desensitization may also be mediated by second-messenger feedback. For example,
β
adrenoceptors stimulate cAMP accumulation, which leads to activation of protein
kinase A; protein kinase A can phosphorylate residues on β receptors, resulting
in inhibition of receptor function. For the β2 receptor, phosphorylation occurs on serine
residues both in the third cyto-plasmic loop and in the carboxyl terminal tail
of the receptor. Similarly, activation of protein kinase C by Gq-coupled
receptors may lead to phosphorylation of this class of G protein-coupled
receptors. This second-messenger feedback mechanism has been termed
heterologous desensitization because activated protein kinase A or protein
kinase C may phosphorylate any structurally similar receptor with the
appropriate consensus sites for phospho-rylation by these enzymes.
Adrenoceptor Polymorphisms
Since
elucidation of the sequences of the genes encoding the α1, α2, and β subtypes of adrenoceptors, it has become clear
that thereare relatively common genetic polymorphisms for many of these
receptor subtypes in humans. Some of these may lead to changes in critical
amino acid sequences that have pharmacologic impor-tance. Often, distinct
polymorphisms occur in specific combina-tions termed haplotypes. Some polymorphisms have been shown to alter
susceptibility to diseases such as heart failure, others to alter the
propensity of a receptor to desensitize, and still others to alter therapeutic
responses to drugs in diseases such as asthma. This remains an area of active
research because studies have reported inconsistent results as to the pathophysiologic
impor-tance of some polymorphisms.
The Norepinephrine Transporter
When
norepinephrine is released into the synaptic cleft, it binds to postsynaptic
adrenoceptors to elicit the expected physiologic effect. However, just as the
release of neurotransmitters is a tightly regulated process, the mechanisms for
removal of neurotransmit-ter must also be highly effective. The norepinephrine
transporter (NET) is the principal route by which this occurs. It is
particularly efficient in the synapses of the heart, where up to 90% of
released norepinephrine is removed by the NET. Remaining synaptic
nor-epinephrine may escape into the extrasynaptic space and enter the
bloodstream or be taken up into extraneuronal cells and metabo-lized by
catecholamine-N-methyltransferase. In
other sites such as the vasculature, where synaptic structures are less well
developed, removal may still be 60% or more by NET. The NET, often situ-ated on
the presynaptic neuronal membrane, pumps the synaptic norepinephrine back into the
neuron cell cytoplasm. In the cell, this norepinephrine may reenter the
vesicles or undergo metabo-lism through monoamine oxidase to
dihydroxyphenylglycol

(DHPG).
Elsewhere in the body similar transporters remove dop-amine (dopamine
transporter, DAT), serotonin (serotonin trans-porter, SERT), and other
neurotransmitters. The NET, surprisingly, has equivalent affinity for dopamine
as for norepinephrine, and it can sometimes clear dopamine in brain areas where
DAT is low, like the cortex.
Blockade
of the NET, eg, by the nonselective psychostimulant cocaine or the NET
selective agents atomoxetine or reboxetine, impairs this primary site of
norepinephrine removal and thus synaptic norepinephrine levels rise, leading to
greater stimulation of α and β adrenoceptors. In the periphery this effect
may pro-duce a clinical picture of sympathetic activation, but it is often
counterbalanced by concomitant stimulation of α2 adrenoceptors in the brainstem that reduces
sympathetic activation.
However,
the function of the norepinephrine and dopamine transporters is complex, and
drugs can interact with the NET to actually reverse the direction of transport
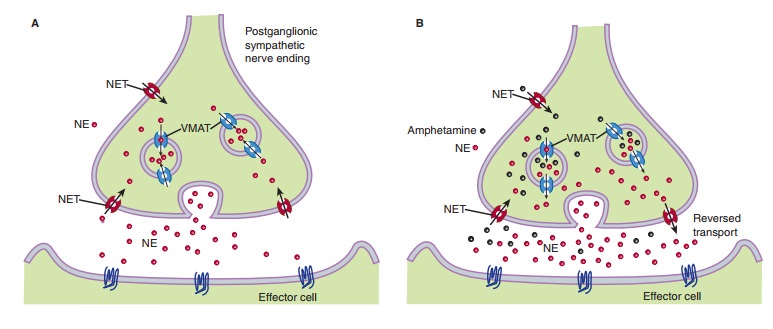
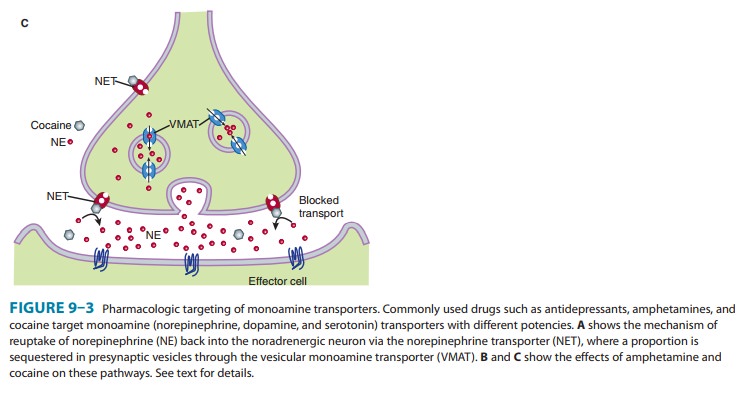
and induce the release of intraneuronal neurotransmitter. This is illustrated
in Figure 9–3. Under normal circumstances (panel A), presynaptic NET (red)
inactivates and recycles norepinephrine (NE, red) released by vesicular fusion.
In panel B, amphetamine (black) acts as both an NET substrate and a reuptake
blocker, eliciting reverse transport and blocking normal uptake, thereby
increasing NE levels in and beyond the synaptic cleft. In panel C, agents such
as methylpheni-date and cocaine (hexagons) block NET-mediated NE reuptake and
enhance NE signaling.
Related Topics