Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Principles of Laboratory Diagnosis of Infectious Diseases
Methods for Detecting an Antigen-Antibody Reaction
IMMUNOLOGIC SYSTEMS
Diagnostic microbiology makes great use of the specificity of the binding between antigen and antibody. Antisera of known specificity are used to detect their homologous antigen in cultures, or more recently, directly in body fluids. Conversely, known antigen preparations are used to detect circulating antibodies as evidence of a current or previous infection with that agent. Many methods are in use to demonstrate the antigen – antibody binding. The greatly improved specificity of monoclonal antibodies has had a major impact on the quality of methods where they have been applied. Before discussing their application to di-agnosis, the principles involved in the most important methods will now be discussed.
Methods for Detecting an Antigen–Antibody Reaction
Precipitation
When antigen and antibody combine in the proper proportions, a visible precipitate is formed (Fig 15 – 9A). Optimum antigen – antibody ratios can be produced by allowing one to diffuse into the other, most commonly through an agar matrix (immunodiffusion).
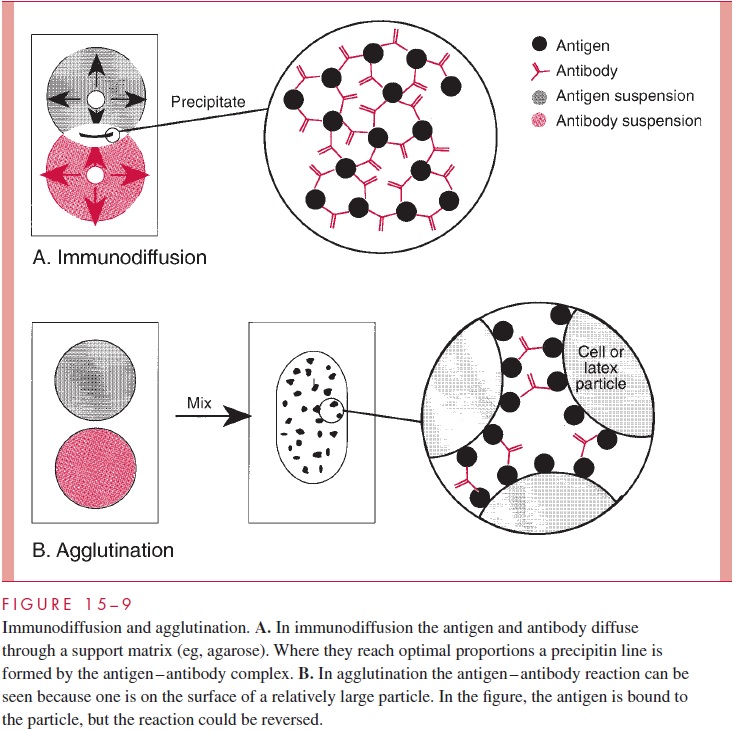
In the immunodiffusion procedure, wells are cut in the agar and filled with antigen and antibody. One or more precipitin lines may be formed between the antigen and antibody wells; depending on the number of different antigen–antibody reactions occurring. Counterimmunoelectrophoresis (CIE) is immunodiffusion carried out in an electrophoretic field. The net effect is that antigen and antibody are rapidly brought together in the space between the wells to form a precipitin line.
Agglutination
The amount of antigen or antibody necessary to produce a visible immunologic reaction can be reduced if either is on the surface of a relatively large particle. This condition can be pro-duced by fixing soluble antigens or antibody onto the surface of RBCs or microscopic latex particles (Fig 15 – 9B). Whole bacteria are large enough to serve as the particle if the anti-gen is present on the microbial surface. The relative proportions of antigen and antibody thus become less critical, and antigen – antibody reactions are detectable by agglutination when immune serum and particulate antigen, or particle-associated antibody and soluble antigen, are mixed on a slide. The process is termed bacterial agglutination, passive hemag-glutination, or latex agglutination depending on the nature of the sensitized particle.
Neutralization
Neutralization as commonly used takes some observable function of the agent, such as cy-topathic effect of viruses or the action of a bacterial toxin, and neutralizes it. This is usu-ally done by first reacting the agent with antibody, and then placing the antigen – antibody mixture into the test system. The steps involved are illustrated in Figure 15 – 10. In viral
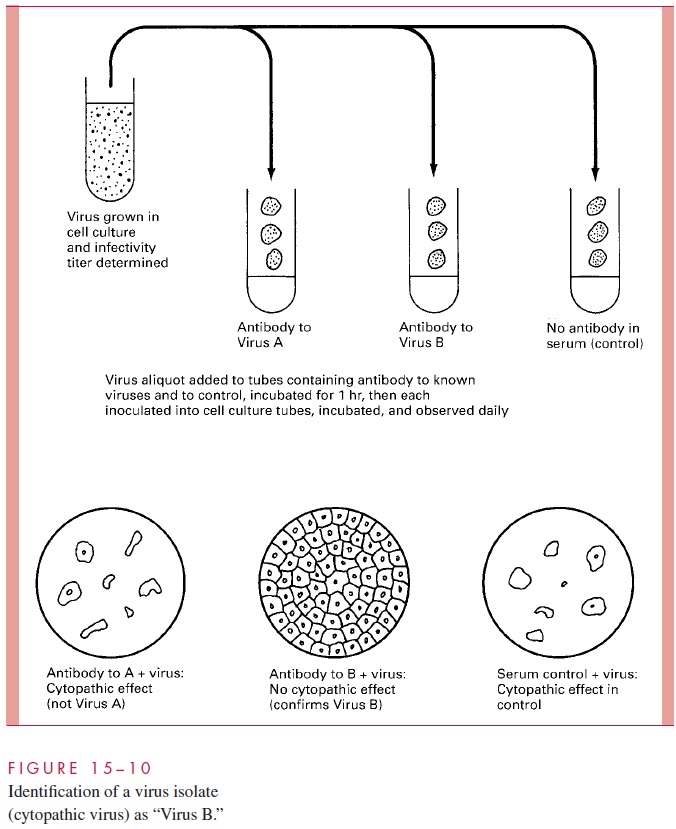
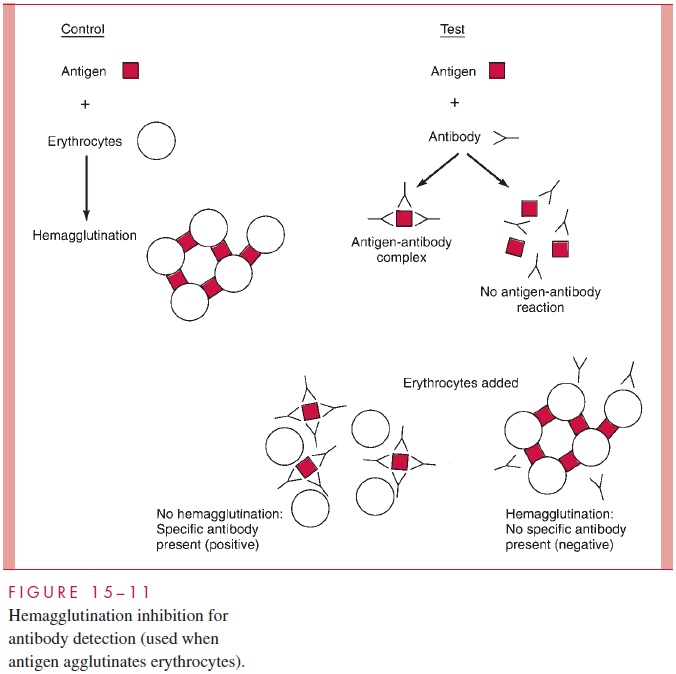
neutralization, a single antibody molecule can bind to surface components of the extracel-lular virus and interfere with one of the initial events of the viral multiplication cycle (adsorption, penetration, or uncoating). Some bacterial and viral agents directly bind to RBCs (hemagglutination). Neutralization of this reaction by antibody blocking of the re-ceptor is called hemagglutination inhibition (Fig 15 – 11).
Complement Fixation
Complement fixation assays depend on two properties of complement. The first is fixation (inactivation) of complement on formation of antigen – antibody complexes. The second is the ability of bound complement to cause hemolysis of sheep (RBCs coated with anti-sheep RBC antibody (sensitized RBCs). Complement fixation assays are performed in two stages: The test system reacts the antigen and antibody in the presence of comple-ment; the indicator system, which contains the sensitized RBCs, detects residual comple-ment. Hemolysis indicates that complement was present in the indicator system and therefore that antigen – antibody complexes were not formed in the test system. Primarily used to detect and quantitate antibody, complement fixation is gradually being replaced by simpler methods.
Labeling Methods
Detection of antigen – antibody binding may be enhanced by attaching a label to one (usu-ally the antibody) and detecting the label after removal of unbound reagents. The label may be a fluorescent dye (immunofluorescence), a radioisotope (radioimmunoassay, or RIA), or an enzyme (enzyme immunoassay, or EIA). The presence or quantitation ofantigen – antibody binding is measured by fluorescence, radioactivity, or the chemical re-action catalyzed by the enzyme.
Immunofluorescence The most common labeling method in diagnostic microbiology isimmunofluorescence (Fig 15 – 12), in which antibody labeled with a fluorescent dye, usu-allyfluorescein isothiocyanate (FITC), is applied to a slide of material that may contain the antigen sought.
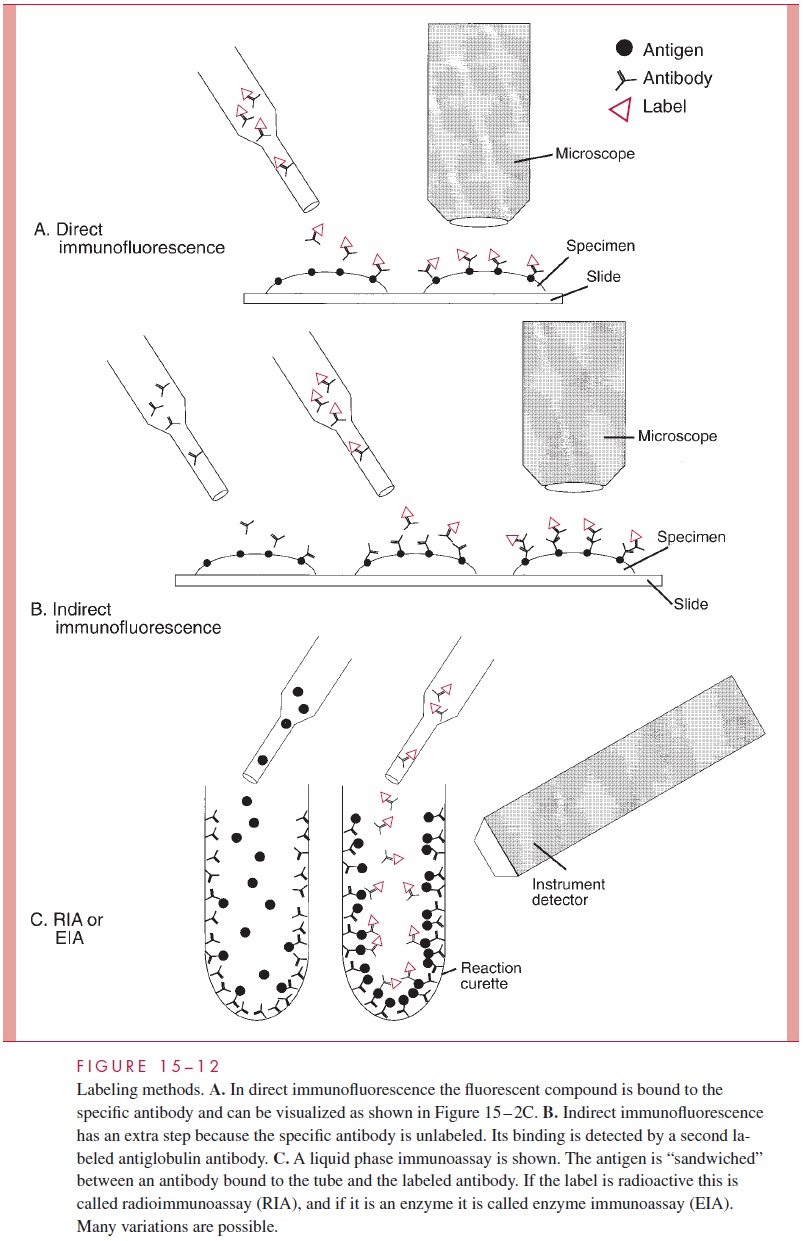
Under fluorescence microscopy, binding of the labeled antibody can be detected as a bright green halo surrounding bacterium, or in the case of viruses, as a fluores-cent clump or on an infected cell. The method is called “direct” if the FITC is conjugated directly to the antibody with the desired specificity. In “indirect” immunofluorescence the specific antibody is not labeled, but its binding to an antigen is detected in an additional step using an FITC-labeled anti-immunoglobulin antibody that will bind to the specific antibody. Choice between the two approaches involves purely technical considerations.
Radioimmunoassay (RIA) and Enzyme Immunoassay (EIA) The labels used in RIAand EIA are more suitable for liquid phase assays and are particularly used in virology. They are also used in direct and indirect methods and many other ingenious variations such as the “sandwich” methods, so called because the antigen of interest is “trapped” be-tween two antibodies (Fig 15 – 12C). These extremely sensitive techniques will be dis-cussed further with regard to antibody detection.
Related Topics