Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Biophysical and Biochemical Analysis of Recombinant Proteins
Immunoassays ELISA - Proteins Analytical Techniques
Immunoassays ELISA
Enzyme-linked immunosorbent assay (ELISA) pro-vides a means to
quantitatively measure extremely small amounts of proteins in biological fluids
and serves as a tool for analyzing specific proteins during purification. This
procedure takes advantage of the observation that plastic surfaces are able to
adsorb low but detectable amounts of proteins. This is a solid phase assay.
Therefore, antibodies against a certain desired protein are allowed to adsorb
to the surface of microtitration plates. Each plate may contain up to 96 wells
so that multiple samples can be assayed. After incubating the antibodies in the
wells of the plate for a specific period of time, excess antibody is removed
and residual protein binding sites on the plastic are blocked by incubation
with an inert protein. Several microtitration plates can be prepared at one
time since the antibodies coating the plates retain their binding capacity for
an extended period. During the ELISA, sample solution containing the protein of
interest is incubated in the wells and the protein (Ag) is captured by the
antibodies coating the well surface. Excess sample is removed and other
antibodies which now have an enzyme (E) linked to them are added to react with
the bound antigen.
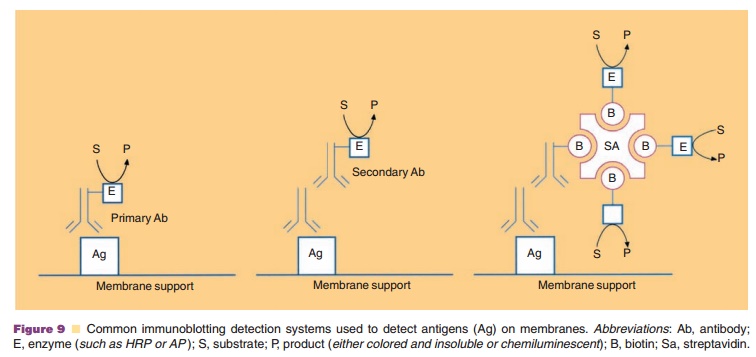
The format described above is called a sandwich assay since the antigen
of interest is located between the antibody on the titer well surface and the
antibody containing the linked enzyme. Figure 10 illustrates anumber of formats
that can be used in an ELISA. A suitable substrate is added and the enzyme
linked to the antibody–antigen–antibody well complex con-verts this compound to
a colored product. The amount of product obtained is proportional to the enzyme
adsorbed in the well of the plate. A standard curve can be prepared if known
concentrations of antigen are tested in this system, and the amount of antigen
in unknown samples can be estimated from this standard curve. A number of
enzymes can be used in ELISAs. However, the most common ones are HRP and AP. A
variety of substrates for each enzyme are available which yield colored
products when catalyzed by the linked enzyme. Absorbance of the colored product
solutions is measured on plate readers, instruments which rapidly measure the
absorbance in all 96 wells of the microtitration plate, and data processing can
be automated for rapid throughput of information. Note that detection
approaches partly parallel those discussed in the section on Blotting. The
above ELISA format is only one of many different methods. For example, the
microtitration wells may be coated directly with the antigen rather than having
a specific antibody attached to the surface. Quantitation is made by comparison
with known quantities of antigen used to coat individual wells.
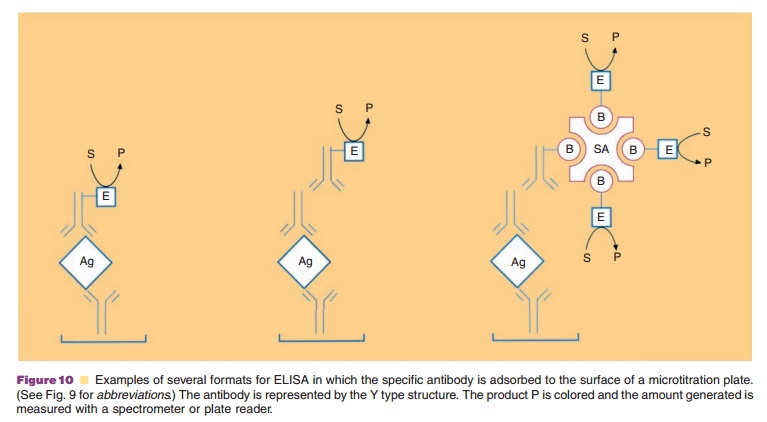
Another approach, this time subsequent to the binding of antigen either
directly to the surface or to an antibody on the surface, is to use an antibody
specific to the antibody binding the protein antigen,that is, a secondary
antibody. This latter, secondary, antibody contains the linked enzyme used for
detec-tion. As already discussed in the section on blotting, the advantage to
this approach is that such antibodies can be obtained in high purity and with
the desired enzyme linked to them from commercial sources. Thus, a single
source of enzyme-linked antibody can be used in assays for different protein
antigens. Should a sandwich assay be used, then antibodies from different
species need to be used for each side of the sandwich. A possible scenario is
that rabbit antibodies are used to coat the microtitration wells; mouse
antibodies, possibly a monoclonal antibody, are used to complex with the
antigen and then a goat anti-mouse immunoglobulin containing linked HRP or AP
is used for detection purposes.
As with immunoblots discussed above, strepta-vidin or avidin can be used
in these assays if biotin is covalently linked to the antibodies and enzymes
(Fig. 10).
If a radioactive label is used in place of the enzyme in the above
procedure, then the assay is a solid phase radioimmunoassay (RIA). Assays are
moving away from the use of radioisotopes, because of problems with safety and
disposal of radioactive waste and since non-radioactive assays have compar-able
sensitivities.
Related Topics