Chapter: Medical Surgical Nursing: Management of Patients With HIV Infection and AIDS
HIV Infection and AIDS: Pathophysiology
Pathophysiology
Since
HIV infection is an infectious disease, it is important to un-derstand how HIV
integrates itself into a person’s immune sys-tem and how immunity plays a role
in the course of infection. This knowledge is also essential for understanding
drug therapy and vaccine development.
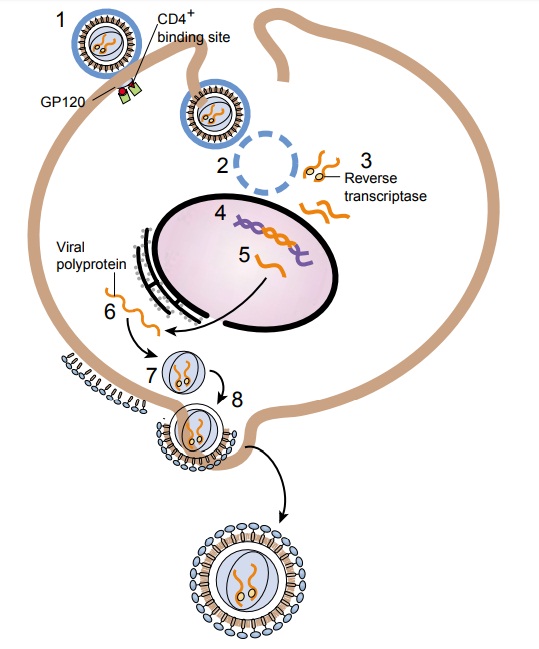
Viruses are intracellular parasites. HIV belongs to a group of viruses known as retroviruses. These viruses carry their genetic material in the form of ribonucleic acid (RNA) rather than de-oxyribonucleic acid (DNA). As can be seen in Figure 52-1, HIV consists of a viral core containing the viral RNA that is sur-rounded by an envelope consisting of glycoproteins (gp) that pro-trude. For HIV to enter the targeted cell, the membrane of the viral envelope must be fused with the plasma membrane of the cell, a process mediated by the envelope glycoproteins of HIV (Wyatt & Sodroski, 1998).
The HIV life cycle is complex and consists of a number of steps (Fig. 52-2). First, the HIV GP120 and GP41 attach to the uninfected CD4 cell surface (receptor) and fuse with the cell membrane.
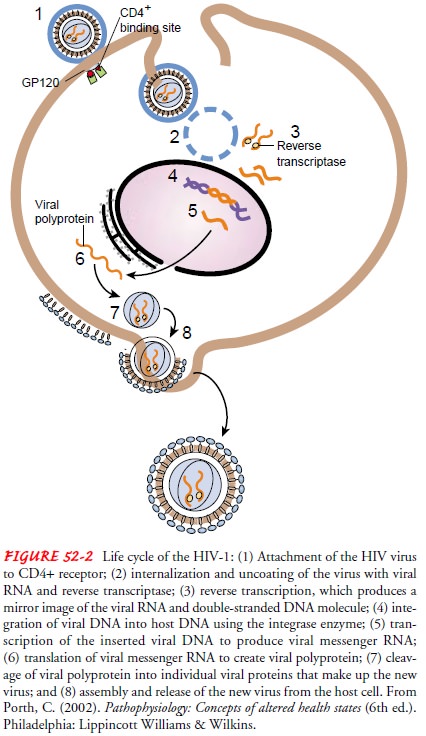
Second, the viral core contents are
emptied into the host cell, a process known as uncoating. Third, HIV enzyme re-verse transcriptase copies the viral
genetic material from RNAinto double-stranded DNA. Fourth, double-stranded DNA
is spliced into the cellular DNA by the action of another HIV en-zyme
integrase. Fifth, using the integrated DNA or provirus as a blueprint, the cell makes new viral proteins and
viral RNA. Sixth, HIV protease cleaves the new proteins (polyproteins).
Seventh, the new proteins join the viral RNA into new viral particles. Finally,
new viral particles bud from the cell and start the process all over (Porth,
2002).
Perhaps the least understood aspects of the
HIV life cycle are the events that occur immediately after entry of the viral
core into the host CD4 cell. The process of viral assembly is intimately linked
to its subsequent reversal (disassembly). Interactions occur between viral
proteins and cellular factors in the cells that produce virus and frequently
influence events at the next round of infection (Emerman & Malim, 1998). In
resting (nondividing) cells, HIV can apparently survive in a latent state as an
integrated provirus that produces few or no viral particles. These resting CD4
T cells can be prodded to churn out new particles if something reactivates them (Mellors, 1998).
When a T cell that harbors this integrated DNA (also
known as provirus) becomes activated against HIV or other microbes, the cell replicates
and also unwittingly begins to produce new copies of both RNA and viral
proteins (Bartlett & Moore, 1998). Activation of the infected cell may be
achieved by antigens, mitogens, select cytokines (tumor necrosis factor-alpha
or interleukin-1), or virus gene products of such viruses as cyto-megalovirus
(CMV), Epstein-Barr virus, herpes simplex virus, and hepatitis. Consequently,
whenever the infected T4 cell is ac-tivated, HIV replication and budding occur,
which often destroys the host cell. Newly formed HIV is then released into the
blood plasma and infects other CD4 cells (see Fig. 52-1).
HIV-1
mutates quickly, at a relatively constant rate, with about 1% of the virus’s
genetic material changing annually. Over the years, 10 distinct subtypes of HIV-1
have emerged, desig-nated A through J. Subtype B is the dominant subtype in
North America and Europe, while subtype D predominates in Africa (Stephenson,
1998).
All viruses target specific cells.
Lymphocytes consist of three major populations: T cells, B cells, and natural
killer cells (Hus-ton, 1997). Mature T cells are phenotypically composed of two
major subpopulations defined by cell surface reciprocal expres-sion of CD4 or
CD8 (Huston, 1997). Approximately two thirds of peripheral blood T cells are
CD4 and approximately one third are CD8. Most people have about 700 to 1,000
CD4 cells/mm3,
but as low as 500/mm3 can be considered “normal.” HIV targets cells with the CD4
glycoprotein, which is expressed on the sur-face of T lymphocytes, monocytes, dendritic cells, and brain
mi-croglia (Wyatt & Sodroski, 1998). A major function of CD4 binding is to
induce conformational changes in the GP120 glyco-protein of the HIV envelope
that contribute to the formation or exposure of the binding site for the chemokine
receptors (Wyatt Sodroski, 1998). Most primary clinical isolates of HIV use the
chemokine receptor CCR5 for entry.
HIV-1 isolates arising later in the course of infection often use other
chemokine receptors such as CXCR4 in addition to CCR5 (Wyatt & Sodroski,
1998). HIV must attach to both the CD4 and CCR5 binding sites in order to
infect CD4+ cells. CD4 fits into a recessed pocket in the viral envelope GP120 that
may simply be too deep to be easily ac-cessed by antibodies (Balter, 1998). A
mutation of CCR5 was identified that is common among Caucasians but not other
ethnic groups. About 1% of Caucasians lack functional CCR5 and are highly
protected against HIV infection even if exposed (although protection is not
absolute); about 18% are not markedly pro-tected against infection but if
infected demonstrate significantly slower rates of disease progression
(Collman, 1997).
Related Topics