Chapter: Organic Chemistry: Aromatic chemistry
Electrophilic substitutions of mono-substituted aromatic rings
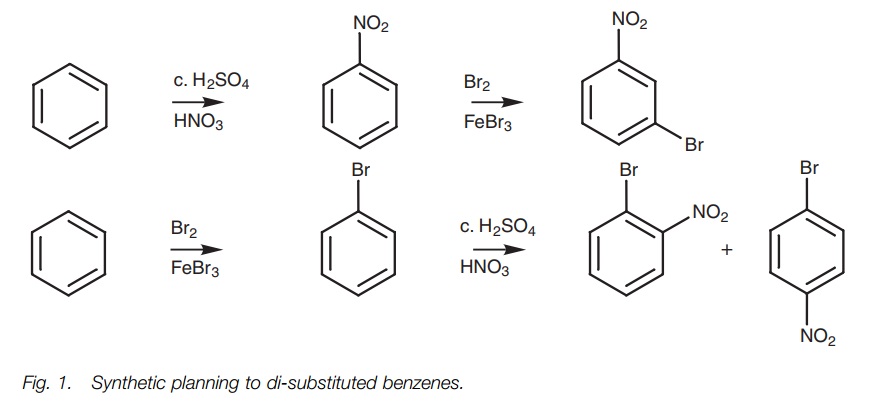
ELECTROPHILIC SUBSTITUTIONS OF MONO-SUBSTITUTED
AROMATIC RINGS
Key Notes
ortho, meta and para substitution
Electrophilic
substitution can occur at three different positions on a mono-substituted
aromatic ring. These positions are defined as the ortho, meta, and para positions.
Substituent effect
Substituents
on an aromatic ring can activate or deactivate the ring towards further
electrophilic substitution. They can also direct substitution either to the meta position or to the ortho and para positions. Inductive and resonance effects determine the
directing effect. Substituents can be divided into four groups depending on
whether they are activating or deactivating, and on their directing effect.
Reaction profile
The
first step of electrophilic substitution is the rate-determining step. The ease
with which electrophilic substitution takes place is determined by the
activation energy of this step, which depends on the stability of the
transi-tion state. The transition state resembles the carbocation intermediate;
and so factors stabilizing the carbocation also stabilize the transition state
and lower the activation energy.
Activating groups – inductive o/p directing
Alkyl
substituents activate aromatic rings towards electrophilic substitution because
they have an inductive effect which pushes electron density into the ring. This
increases the nucleophilicity of the ring towards electrophiles and also
stabilizes the positively charged intermediate; ortho/para attack is
favored over meta attack.
Deactivating groups – inductive m directing
Inductive
deactivating groups are groups having an electron deficient atom directly
attached to the aromatic ring. They include substituents such as nitro, acyl,
carboxylic acid, sulfonic acid, and nitrile groups. Such groups have an
electron-withdrawing effect which makes the aromatic ring less nucleophilic and
less reactive to electrophiles; meta
attack is favored over ortho/para.
Activating groups – resonance o/p directing
Substituents
such as phenols, ethers, amines, and amides activate the aro-matic ring and
direct substituents to the ortho and para positions. In all these
substituents, a nitrogen or an oxygen with a lone pair of electrons is attached
directly to the ring and interacts with the ring by resonance. This resonance
effect outweighs any electron-withdrawing effect of the elec-tronegative oxygen
or nitrogen. Amides are less activating than amines. This can be useful since
amines can be temporarily converted to amides in order to lower reactivity and
direct electrophilic substitution to the para
position.
Deactivating groups – resonance o/p directing
Halogen
substituents deactivate aromatic rings towards electrophilic sub-stitution by
an inductive effect. These atoms are strongly electronegative and have an
electron-withdrawing effect on the ring making it less nucleo-philic and less
reactive. However, once electrophilic substitution does take place, halogens
direct to the ortho and para positions due to a resonance effect
which helps to delocalize the positive charge onto the halogen atoms. This
resonance effect is more important than the inductive effect which would
normally direct substitution to the meta
position.
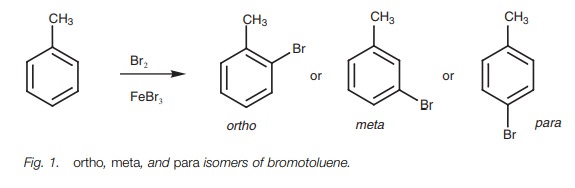
ortho, meta and para substitution
Aromatic compounds which already contain a
substituent can undergo electro-philic substitution at three different
positions relative to the substituent. Consider the bromination of toluene (Fig. 1). Three different products are
possible depending on where the bromine enters the ring. These products have
the same molecular formula and are therefore constitutional isomers. The
aromatic ring is said to be disubstituted and the three possible isomers are
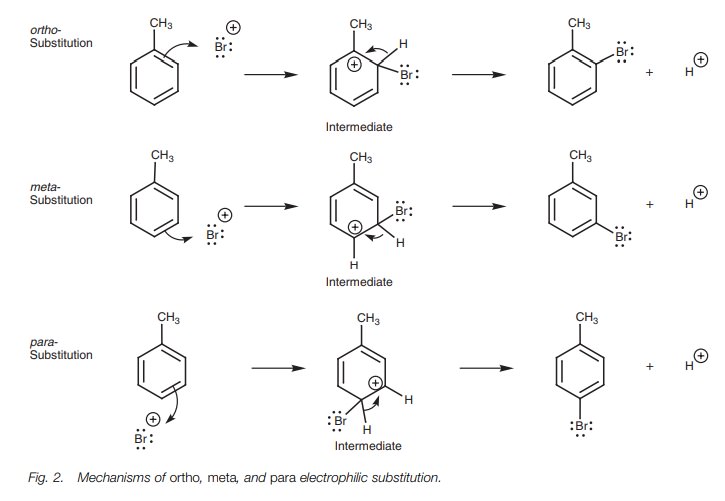
described as being ortho, meta, and para. The mechanisms leading to these three isomers are shown in Fig. 2.
Substituent effect
Of the three possible isomers arising from the
bromination of toluene, only two (the ortho
and para) are formed in significant
quantity. Furthermore, the bromination of toluene goes at a faster rate than
the bromination of benzene. Why? The answer lies in the fact that the methyl
substituent can affect the rate and the position of further substitution. A
substituent can either activate or deactivate the aromatic ring towards
electrophilic substitution and does so through inductive or resonance effects.
A substituent can also direct the next substitution so that it goes mainly ortho/para or mainly meta. We
can classify substituents into four groups depending on the effect they have on
the rate and the position of substitution, that is:
·
activating groups which direct ortho/para by inductive effects;
·
deactivating groups which direct meta by inductive effects;
·
activating groups which direct ortho/para by resonance effects;
·
deactivating groups which direct ortho/para by resonance
effects.
There are no substituents which activate the ring and direct meta.
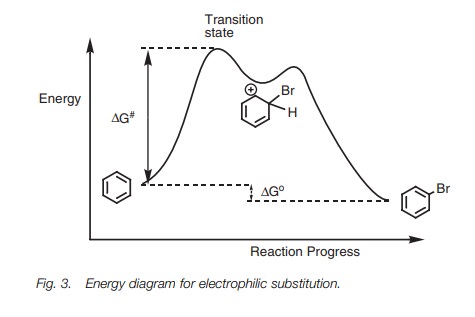
Reaction profile
Before explaining the reasons behind the
substituent effect, we have to consider the reaction profile of electrophilic
substitution with respect to the relative energies of starting material,
intermediate, and product. The energy diagram (Fig.3) illustrates the
reaction pathway for the bromination of benzene. The first stage in the
mechanism is the
rate-determining step and is the
formation of the carbocation. This is endothermic and
proceeds through a transition state which requires an activation energy (∆G# ). The magnitude of ∆G#
determines the rate at which the reaction will occur and this in turn is
determined by the stability of the transition state. The transition state
resembles the carbocation intermediate and so any factor which stabilizes the
intermediate also stabilizes the transition state and favors the reaction.
Therefore, in the discussions to follow we can consider the stability of
relative carbocations in order to determine which reaction is more favorable.
Activating groups – inductive o/p directing
A methyl substituent is an example of an
inductive activating group and so we shall consider again the bromination of
toluene. In order to explain the directing properties of the methyl group, we
need to look more closely at the mechanisms involved in generating the ortho, meta, and para isomers (Fig. 2). The preferred reaction pathway
will be the one which goes through the most stable intermedi- ate. Since a
methyl group directs ortho and para, the intermediates involved in
these reaction pathways are more stable than the intermediate involved in metasubstitution. The relevant
intermediates and their resonance structures are shown in Fig. 4.
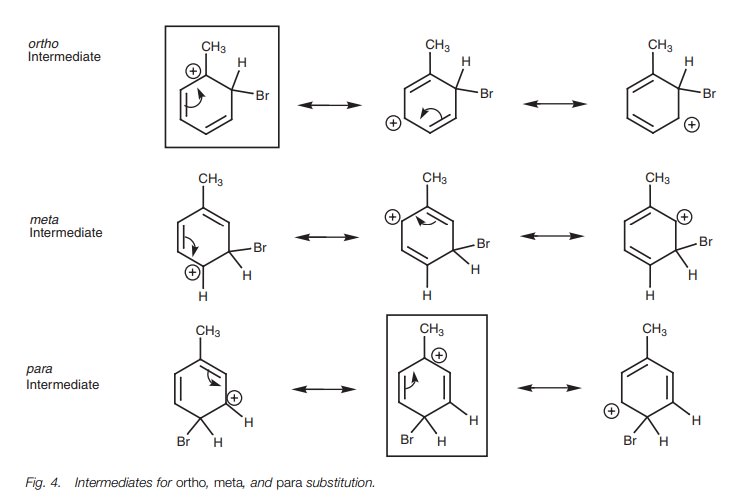
If we compare all the resonance structures above, we can spot one ortho and one para resonance structure (boxed) where the positive charge is positioned immediately next to the methyl substituent.
An
alkyl group can stabilize a neigh-boring positive charge by an inductive,
electron-donating effect which results in some of the positive charge being
spread over the alkyl group. This is an addi-tional stabilizing effect which is
only possible for the intermediates arising from ortho and para substitution.
There is no such equivalent resonance structure for themetaintermediate and so that means that the ortho and para intermediates
experi-ence an increased stability over the meta,
which results in a preference for these two substitution pathways.
By the same token, toluene will be more
reactive than benzene. The electron-donating effect of the methyl group into
the aromatic ring makes the ring inher-ently more nucleophilic and more
reactive to electrophiles, as well as providing extra stabilization of the
reaction intermediate. To sum up, alkyl groups are activating groups and areortho,para directing.
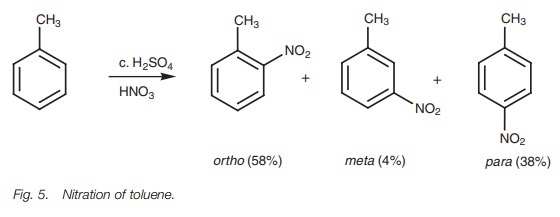
The nitration of toluene also illustrates this effect (Fig. 5). The amount of meta sub-stitution is very small as we would expect, and there is a preference for the ortho and para products. However, why is there more ortho substitution compared to para sub-stitution? Quite simply, there are two ortho sites on the molecule to one para site and so there is double the chance of ortho attack to para attack. Based on pure statistics we would have expected the ratio of ortho to para attack to be 2:1. In fact, the ratio is closer to 1.5:1. In other words, there is lessortho substitution than expected. This is because the ortho sites are immediately ‘next door’ to the methyl substituent and the size of the substituent tends to interfere with ortho attack – a steric effect. The significance of this steric effect will vary according to the size of the alkyl sub-stituent. The larger the substituent, the more ortho attack will be hindered.
Deactivating groups – inductive m directing
Alkyl
groups are activating
groups and direct
substitution to the
ortho, para positions. Electron
withdrawing substituents (Fig. 6) have the opposite effect.
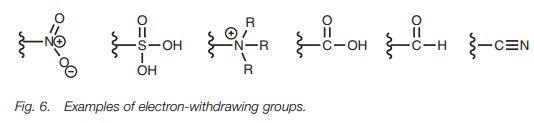
They deactivate the ring, make the ring less nucleophilic and less likely to react with an electrophile. The electron-withdrawing effect also destabilizes the reac- tion intermediate and makes the reaction more difficult. This destabilization is more pronounced in the intermediates arising from ortho/para attack and so meta attack is favored.
All of these groups have a positively charged
atom or an electron deficient atom (i.e. an electrophilic center) directly
attached to the aromatic ring. Since this atom is electron deficient, it has an
electron-withdrawing effect on the ring.
Deactivating groups make electrophilic
substitution more difficult but the reac-tion will proceed under more forcing
reaction conditions. However, substitution is now directed to the meta position. This can be explained by
comparing all the possible resonance structures arising from ortho, meta and para attack. As
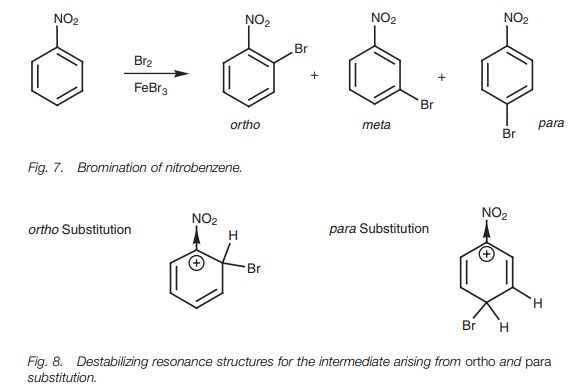
an exam-ple, we shall consider the bromination of nitrotoluene (Fig. 7). Of all the possible resonance
structures arising from ortho, meta, and para attack, there are two spe-cific resonance structures (arising
from ortho and para attack) where the positive charge is placed directly next to
the electron-withdrawing nitro group (Fig.
8). As a result, these resonance structures are greatly destabilized. This
does not occur with any of the resonance structures arising from meta attack and so meta attack is favored.
Activating groups – resonance o/p directing
Phenol
is an example
of a substituent
which activates the
aromatic ring by resonance
effects and which
directs substitution to
the ortho and tions. In phenol, an electronegative
oxygen atom is next to the aromatic ring. Since oxygen is electronegative, it
should have an electron-withdrawing induc- tive effect and so might be expected
to deactivate the ring. The fact that the phenolic group is a powerful
activating group is due to the fact that oxygen is electron
rich and can
also act as
a nucleophile, feeding
electrons into the ring through a resonance process. As an
example, we shall look at the nitra- tion of phenol (Fig. 9).
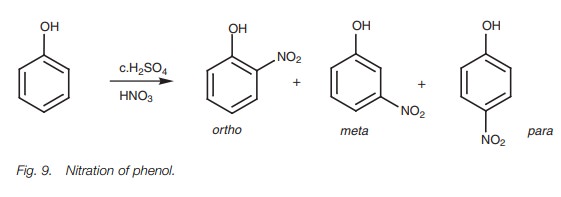
There are three resonance structures for the
intermediate formed in each form of electrophilic substitution, but there are
two crucial ones to consider (Fig. 10),
aris-ing from ortho and para substitution. These resonance
structures have the positive

If oxygen
only had an inductive effect, these resonance structures would be highly
unstable. However, oxygen can act as a nucleophile and can use one of its lone
pairs of electrons to form a new π bond to the neighboring electrophilic center (Fig. 11). This results in a fourth
resonance structure where the positive charge is moved out of the ring and onto
the oxygen atom. Delocalizing the charge like this further stabilizes it and
makes the reaction proceed more easily.
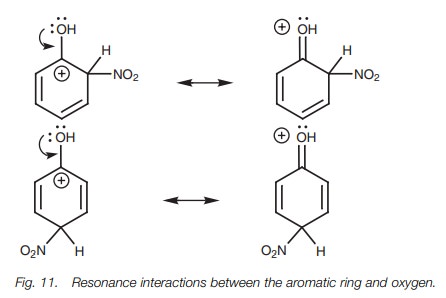
Since none of the resonance structures arising
from meta attack places the posi-tive
charge next to the phenol group, this fourth resonance structure is not
avail-able to the meta intermediate
and so meta attack is not favored.
Thus, the phenol group is an activating group which is ortho, para directing
because of resonance effects. This resonance effect is more important than any
inductive effect which the oxygen might have.
The same holds true for the following
substituents: alkoxy (–OR), esters (–OCOR), amines (–NH2, –NHR, –NR2),
and amides (–NHCOR). In all these cases, there is either a nitrogen or an
oxygen next to the ring. Both these atoms are nucleo-philic and have lone pairs
of electrons which can be used to form an extra bond to the ring. The ease with
which the group can do this depends on the nucleophilicity of the attached atom
and how well it can cope with a positive charge.
Nitrogen is more nucleophilic than oxygen since
it is better able to cope with the resulting positive charge. Therefore amine
substituents are stronger activating groups than ethers. On the other hand, an
amide group is a weaker activating group since the nitrogen atom is less
nucleophilic. This is because the nitrogen’s lone pair of electrons is pulled
towards the carbonyl group and is less likely to form a bond to the ring (Fig. 12). This property of amides can be
quite
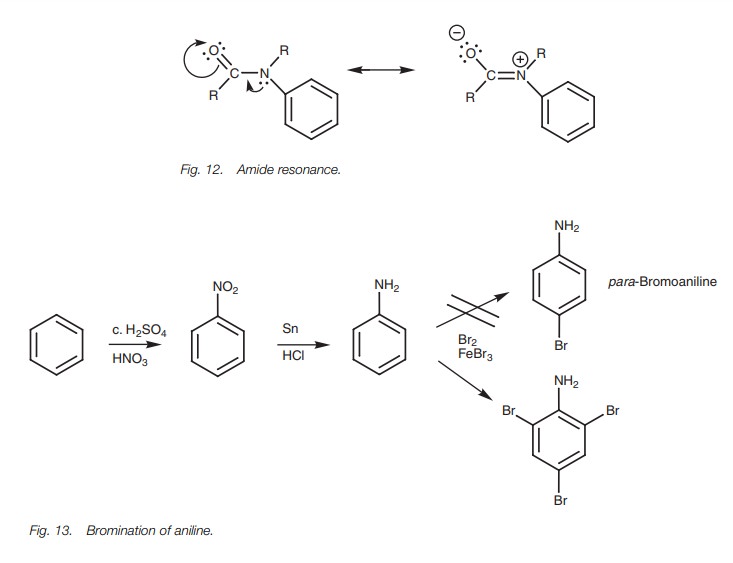
Suppose for example we wanted to make para-bromoaniline by bromi-nating
aniline (Fig. 13). In theory, this
reaction scheme should give the desired product. In practice, the NH2
group is such a strong activating group that the final bromination goes three
times to give the tri-brominated product rather than the mono-brominated
product.
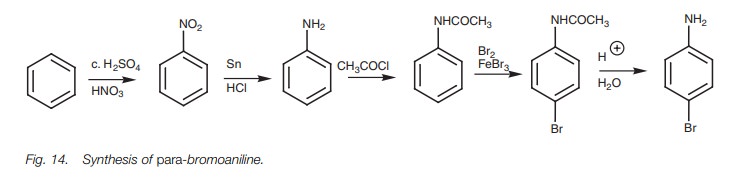
In order to lower the activation of the amino
group, we can convert it to the less activating amide group. The bromination then
only goes once. We also find that the bromination reaction is more selective
for the para position than for the ortho position. This is because the
amide group is bulkier than the NH2 group and tends to shield the ortho positions from attack. Once the
bromination has been completed the amide can be converted back to the amino
group by hydrolysis.
Deactivating groups – resonance o/p directing
The fourth and last group of aromatic
substituents are the halogen substituents which deactivate the aromatic ring
and which direct substitution to the ortho and para positions. These are
perhaps the trickiest to understand since they deactivate the ring by one
effect, but direct substitution by a different effect. The halogen atom is
strongly electronegative and therefore we would expect it to have a strong
electron-withdrawing inductive effect on the aromatic ring. This would make the
aromatic ring less nucleophilic and less reactive to electrophiles. It would
also destabilize the required intermediate for electrophilic substitution.
Halogens are also poorer nucleophiles and so any resonance effects they might
have are less important than their inductive effects.
However,
if halogen atoms
are deactivating the
ring because of
inductive effects, why do they not direct substitution to the meta
position like other electron- withdrawing groups? Let us look at a specific
reaction – the nitration of bromo- benzene
(Fig. 15). There are
three resonance structures
for each of
the three intermediates leading
to these products, but the crucial ones to consider are those which position a
positive charge next to the substituent. These occur with ortho and para
substitution, but not meta substitution (Fig. 16). These are the crucial res-
onance structures as far as the directing properties of the substituent is
concerned.
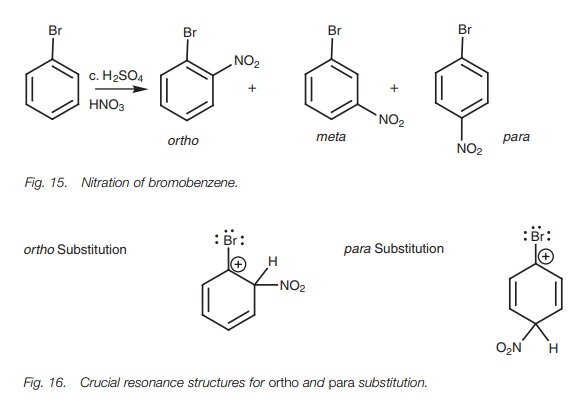
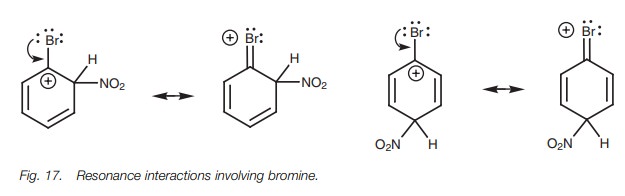
If bromine acts inductively, it will destabilize these intermediates and direct sub- stitution to the meta position. However, we know that bromine directs ortho/para and so it must be stabilizing the ortho/para intermediates rather than destabilizing them. The only way that bromine can stabilize the neighboring positive charge is by resonance in the same way as a nitrogen or oxygen atom (Fig. 17). Thus, the bromine acts as a nucleophile and donates one of its lone pairs to form a new bond to the electrophilic center beside it. A new π bond is formed and the positive charge is moved onto the bromine atom. This resonance effect is weak since the halogen atom is a much weaker nucleophile than oxygen or nitrogen and is less capable of stabilizing a positive charge. However, it is significant enough to direct substitution to the ortho and para positions.
For halogen substituents, the inductive effect
is more important than the reso-nance effect in deactivating the ring. However,
once electrophilic substitution does take
place, resonance effects are more important than inductive effects indirecting
substitution.
Related Topics