Chapter: Basic & Clinical Pharmacology : Skeletal Muscle Relaxants
Basic Pharmacology of Neuromuscular Blocking Drugs
BASIC
PHARMACOLOGY OF NEUROMUSCULAR BLOCKING DRUGS
Chemistry
All of the available neuromuscular blocking drugs bear a
structural resemblance to acetylcholine. For example, succinylcholine is two
acetylcholine molecules linked end-to-end (Figure 27–3).
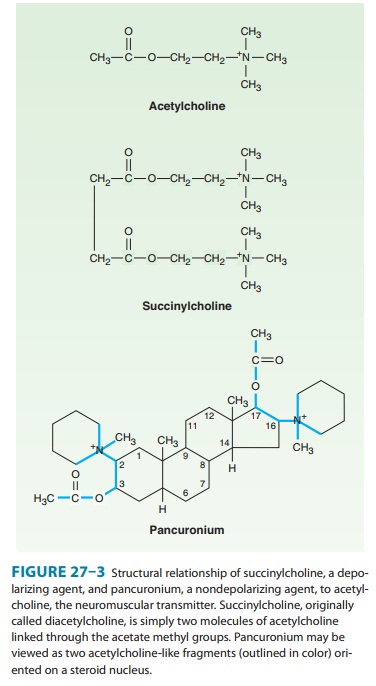
In contrast to
the single linear structure of succinylcholine and other depolarizing drugs,
the nondepolarizing agents (eg, pancuronium) conceal the “double-acetylcholine”
structure in one of two types of bulky, semi-rigid ring systems (Figure 27–3).
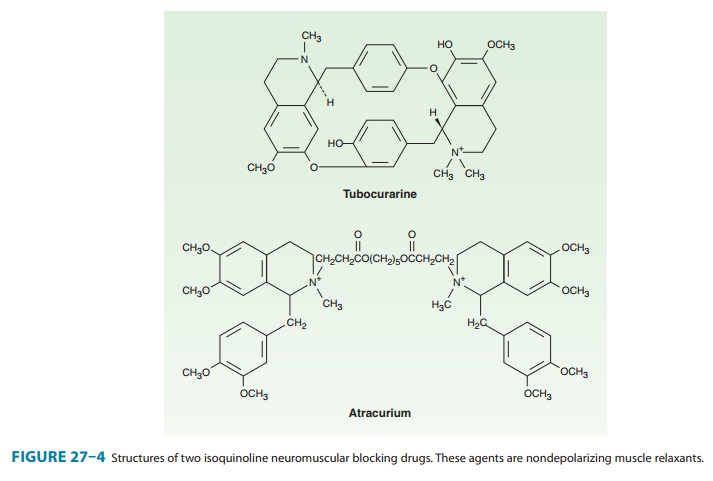
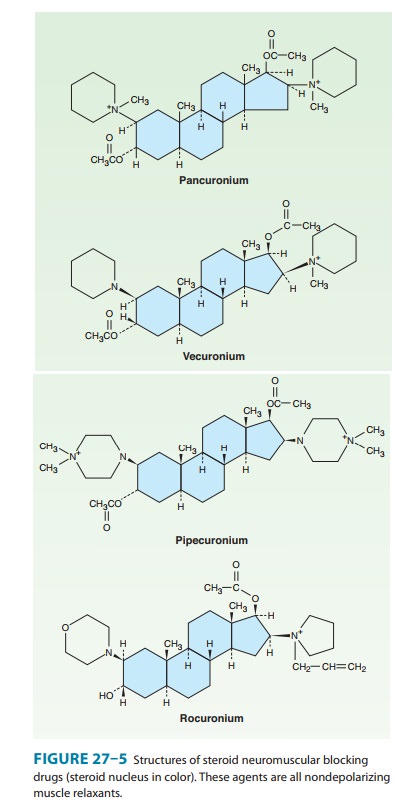
Examples of the two major families of nondepolarizing blocking drugs—the
isoquinoline and steroid derivatives—are shown in Figures 27–4 and 27–5.
Another feature common to all currently used neuromuscular blockers is the
presence of one or two quaternary nitrogens, which makes them poorly lipid
soluble and limits entry into the CNS.
Pharmacokinetics of Neuromuscular Blocking Drugs
All
of the neuromuscular blocking drugs are highly polar com-pounds and inactive
orally; they must be administered parenterally.
A. Nondepolarizing Relaxant Drugs
The
rate of disappearance of a nondepolarizing neuromuscular blocking drug from the
blood is characterized by a rapid initial distribution phase followed by a
slower elimination phase. Neuromuscular blocking drugs are highly ionized, do
not readily cross cell membranes, and are not strongly bound in peripheral
tissues. Therefore, their volume of distribution (80–140 mL/kg) is only
slightly larger than the blood volume.
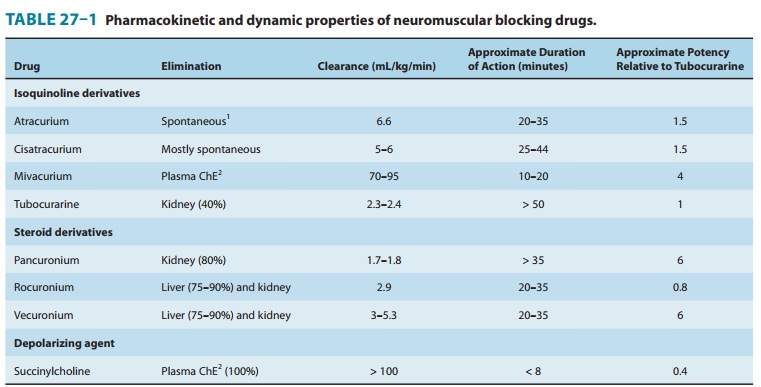
The
duration of neuromuscular blockade produced by nonde-polarizing relaxants is
strongly correlated with the elimination half-life. Drugs that are excreted by
the kidney typically have lon-ger half-lives, leading to longer durations of
action (> 35 minutes). Drugs eliminated by the liver tend to have shorter
half-lives and durations of action (Table 27–1). All steroidal muscle relaxants
are metabolized to their 3-hydroxy, 17-hydroxy, or 3,17-dihydroxy products in
the liver. The 3-hydroxy metabolites are usually 40–80% as potent as the parent
drug. Under normal circum-stances, metabolites are not formed in sufficient
quantities to produce a significant degree of neuromuscular blockade during or
after anesthesia. However, if the parent compound is administered for several
days in the ICU setting, the 3-hydroxy metabolite may accumulate and cause
prolonged paralysis because it has a longer half-life than the parent compound.
The remaining metabolites possess minimal neuromuscular blocking properties.
The
intermediate-acting steroid muscle relaxants (eg, vecuro-nium and rocuronium)
tend to be more dependent on biliaryexcretion or hepatic metabolism for their
elimination. These muscle relaxants are more commonly used clinically than the
long-acting steroid-based drugs (eg, pancuronium).
Atracurium (Figure 27–4) is an intermediate-acting isoquino-line nondepolarizing muscle relaxant. In addition to hepatic metabolism, atracurium is inactivated by a form of spontaneous breakdown known as Hofmann elimination. The main breakdown products are laudanosine and a related quaternary acid, neither of which possesses neuromuscular blocking properties. Laudanosine is slowly metabolized by the liver and has a longer elimination half-life (ie, 150 minutes). It readily crosses the blood-brain barrier, and high blood concentrations may cause seizures and an increase in the volatile anesthetic requirement. During surgical anesthesia, blood levels of laudanosine typically range from 0.2 to 1 mcg/mL; however, with prolonged infusions of atracurium in the ICU, laudanosine blood levels may exceed 5 mcg/mL.
Atracurium
has several stereoisomers, and the potent isomer cisatracurium has become one of the most commonly usedmuscle
relaxants in clinical practice. Although cisatracurium resembles atracurium, it
has less dependence on hepatic inactiva-tion, produces less laudanosine, and is
less likely to release hista-mine. From the clinical perspective, cisatracurium
has all the advantages of atracurium with fewer side effects. Therefore,
cisa-tracurium has largely replaced atracurium in clinical practice.
Mivacurium, another isoquinoline compound, has the
shortestduration of action of all nondepolarizing muscle relaxants (Table
27–1). However, its onset of action is significantly slower than that of
succinylcholine. In addition, the use of a larger dose to speed the onset can
be associated with profound histamine release leading to hypotension, flushing,
and bronchospasm. Clearance of mivacu-rium by plasma cholinesterase is rapid
and independent of the liver or kidney (Table 27–1). However, because patients
with renal fail-ure often have decreased levels of plasma cholinesterase, the short
duration of action of mivacurium may be prolonged in patients with impaired
renal function. Although mivacurium is no longer in widespread clinical use, an
investigational ultra-short-acting isoquinoline nondepolarizing muscle
relaxant, gantacurium, is cur-rently in phase 3 clinical testing.
Gantacurium represents a new class of nondepolarizing
neuro-muscular blockers, called asymmetric mixed-onium chlorofumar-ates. It is
degraded nonenzymatically by adduction of the aminoacid cysteine and ester bond
hydrolysis. Preclinical and clinical data indicate gantacurium has a rapid
onset of effect and predict-able duration of action (very short, similar to
succinylcholine) that can be reversed with edrophonium or administration of
cysteine. At doses above three times the ED95, cardiovascular
adverse effects have occurred, probably due to histamine release. No
broncho-spasm or pulmonary vasoconstriction has been reported at these higher
doses.
B. Depolarizing Relaxant Drugs
The extremely short duration of action of succinylcholine (5–10 minutes) is due to its rapid hydrolysis by butyrylcholinesterase and pseudocholinesterase in the liver and plasma, respectively. Plasma cholinesterase metabolism is the predominant pathway for succinylcholine elimination. Since succinylcholine is more rapidly metabolized than mivacurium, its duration of action is shorter than that of mivacurium (Table 27–1). The primary metabolite of succi-nylcholine, succinylmonocholine, is rapidly broken down to succinic acid and choline. Because plasma cholinesterase has an enormous capacity to hydrolyze succinylcholine, only a small percentage of the original intravenous dose ever reaches the neuromuscular junction. In addition, because there is little if any plasma cholinesterase at the motor end plate, a succinylcholine-induced blockade is terminated by its diffusion away from the end plate into extracellular fluid. Therefore, the circulating levels of plasma cholinesterase influence the duration of action of succinylcholine by determining the amount of the drug that reaches the motor end plate.
Neuromuscular blockade produced by succinylcholine and
mivacurium can be prolonged in patients with an abnormal genetic variant of
plasma cholinesterase. The dibucaine number
is a measure of the ability of a patient to metabolize succinylcholine and can
be used to identify at-risk patients. Under standardized test conditions,
dibucaine inhibits the normal enzyme by 80% and the abnormal enzyme by only
20%. Many genetic variants of plasma cholines-terase have been identified,
although the dibucaine-related variants are the most important. Given the
rarity of these genetic variants, plasma cholinesterase testing is not a
routine clinical procedure.
Mechanism of Action
The
interactions of drugs with the acetylcholine receptor-end plate channel have
been described at the molecular level. Several modes of action of drugs on the
receptor are illustrated in Figure 27–6.
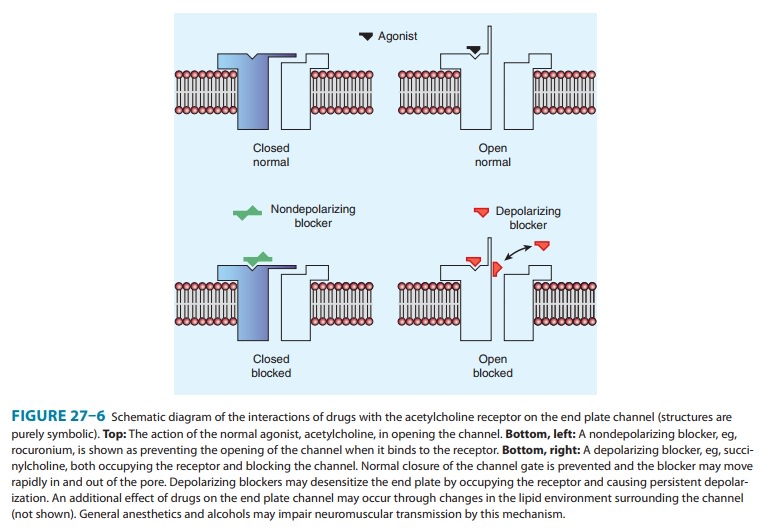
A. Nondepolarizing Relaxant Drugs
All
the neuromuscular blocking drugs in current use in the USA except
succinylcholine are classified as nondepolarizing agents. Although it is no
longer in widespread clinical use, d-tubocurarine is considered the
prototype neuromuscular blocker. When small doses of nondepolarizing muscle
relaxants are administered, they act predominantly at the nicotinic receptor
site by competing with acetylcholine. The least potent nondepolarizing
relaxants (eg, rocuronium) have the fastest onset and the shortest duration of
action. In larger doses, nondepolarizing drugs can enter the pore of the ion
channel (Figure 27–1) to produce a more intense motor blockade. This action
further weakens neuromuscular transmission and diminishes the ability of the
acetylcholinesterase inhibitors (eg, neostigmine, edrophonium, pyridostigmine)
to antagonize the effect of nondepolarizing muscle relaxants.
Nondepolarizing
relaxants can also block prejunctional sodium channels. As a result of this
action, muscle relaxants interfere with the mobilization of acetylcholine at
the nerve ending and cause fade (Figure 27-7). One consequence of the
surmountable nature of the postsynaptic blockade produced by nondepolarizing
muscle relaxants is the fact that tetanic stimulation, by releasing a large
quantity of acetylcholine, is followed by transient posttetanic facilitation of
the twitch strength (ie, relief of blockade). An important clinical consequence
of this principle is the reversal of residual blockade by cholinesterase
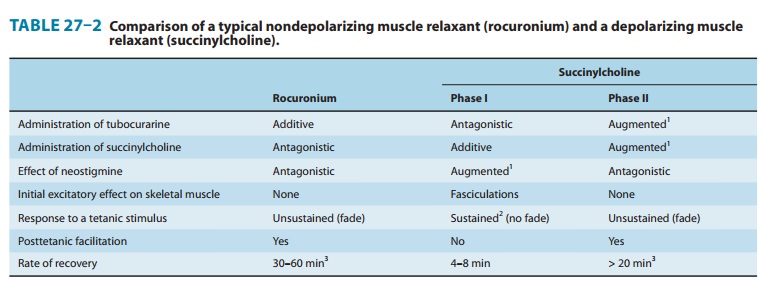
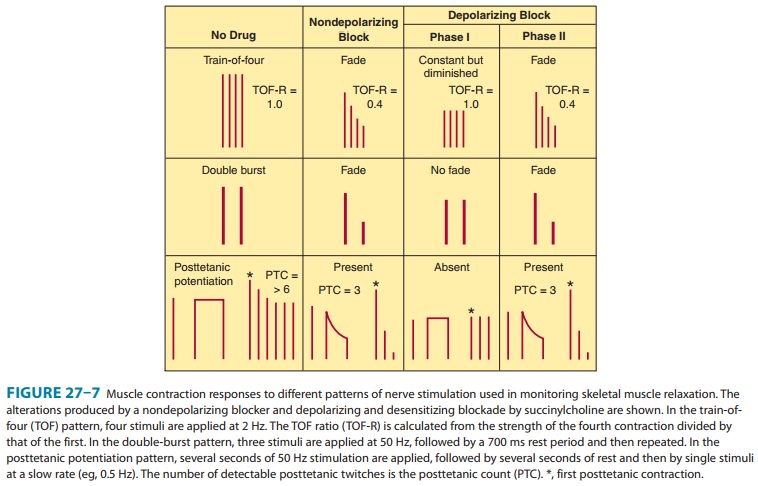
inhibitors. The characteristics of a nondepolarizing neuromuscular blockade are
summarized in Table 27–2 and Figure 27–7.
B. Depolarizing Relaxant Drugs
1. Phase I block (depolarizing)—Succinylcholine is the
onl clinically useful depolarizing blocking drug. Its neuromuscular
effects
are like those of acetylcholine except that succinylcholine produces a longer
effect at the myoneural junction. Succinylcholine
reacts
with the nicotinic receptor to open the channel and causedepolarization of the
motor end plate, and this in turn spreads to the adjacent membranes, causing
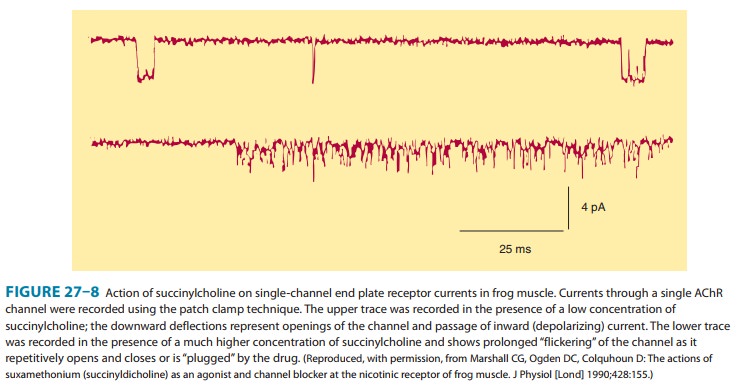
contractions of muscle motor units. Data from single-channel recordings
indicate that depolarizing blockers can enter the channel to produce a
prolonged“flickering” of the ion conductance (Figure 27–8). Because
succinylcholine is not metabolized effectively at the synapse, thedepolarized
membranes remain depolarized and unresponsive tosubsequent impulses(ie,a state
of depolarizingblockade).Furthermore,
because
excitation-contraction
coupling requires end plate repolarization
(“repriming”) and repetitive firing to maintain muscle tension, a flaccid
paralysis results. In contrast to the nondepolarizing drugs, this so-called
phase I (depolarizing) block is augmented, not reversed, by cholinesterase
inhibitors. The characteristics of a depolarizing neuromuscular blockade are
summarized in Table 27–2 and Figure 27–7.
2. Phase II block
(desensitizing)—With prolonged exposureto succinylcholine, the initial end plate
depolarization decreases and the membrane becomes repolarized. Despite this
repolariza-tion, the membrane cannot easily be depolarized again because it is desensitized. The mechanism for this
desensitizing phase is unclear, but some evidence indicates that channel block
may become more important than agonist action at the receptor in phase II of
succinylcholine’s neuromuscular blocking action. Regardless of the mechanism,
the channels behave as if they are in a prolonged closed state (Figure 27–7).
Later in phase II, the char-acteristics of the blockade are nearly identical to
those of a nonde-polarizing block (ie, a nonsustained twitch response to a
tetanic stimulus) (Figure 27–7), with possible reversal by acetylcholines-terase
inhibitors.
Related Topics