Chapter: Medical Immunology: Tolerance and Autoimmunity
Autoimmunity
AUTOIMMUNITY
A. Introduction
Failure of the immune system to “tolerate” self antigens may result in the development of pathological processes known as autoimmune diseases. At the clinical level, autoimmunity is apparently involved in a variety of apparently unrelated diseases such as systemic lupus erythematosus (SLE), insulin-dependent diabetes mellitus, myasthenia gravis, rheumatoid arthritis, multiple sclerosis, and hemolytic anemia. At least 40 diseases are known or con-sidered to be autoimmune in nature, affecting about 5% of the general population. Their distribution by sex and age is not uniform. As a rule, autoimmune diseases predominate in females and have a bimodal age distribution. A first peak of incidence is around puberty, whereas the second peak is in the forties and fifties.
B. Classification of the Autoimmune Diseases
There are several different ways to classify autoimmune diseases. Because several autoim-mune diseases are strongly linked with MHC antigens, one of the most recently proposed classifications, shown in Table 16.5, groups autoimmune diseases according to their asso-ciation with class I or with class II MHC markers. It is interesting to notice that although autoimmune diseases may afflict both men and women, there is female preponderance for the class II–associated diseases and a definite increase in the prevalence of class I–associ-ated diseases among males.

C. Pathophysiology of Autoimmune Diseases
The autoimmune pathological process may be initiated and/or perpetuated by autoantibod-ies, immune complexes containing autoantigens, and autoreactive T lymphocytes. Each of these immune processes plays a preponderant role in certain diseases or may be synergis-tically associated, particularly in multiorgan, systemic autoimmune diseases.
1. The Role of Autoantibodies in Autoimmune Diseases
B lymphocytes with autoreactive specificities remain nondeleted in the adult individuals of many species. In mice, polyclonal activation with lipopolysaccharide leads to production of autoantibodies. In humans, bacterial and viral infections (particularly chronic) may lead to the production of anti-immunoglobulin and antinuclear antibodies. In general, it is ac-cepted that polyclonal B-cell activation may be associated with the activation of autoreac-tive B lymphocytes.
Autoantibody-associated diseases are characterized by the presence of autoantibodies in the individual’s serum and by the deposition of autoantibodies in tissues. The pathogenic role of autoantibodies is not always obvious and depends on several factors, such as the availability and valence of the auto-antigen and the affinity and charge of the antibody.
· Antibodies with high affinity for the antigen are considered to be more pathogenic because they form stable IC that can activate complement more effectively.
· Anti-DNA antibodies of high isoelectric point, very prevalent in SLE, have a weak positive charge at physiological pH and bind to the negatively charged glomerular basement membrane, which also binds DNA. Such affinity of anti-gens and antibodies for the glomerular basement membrane creates the ideal conditions for in situ IC formation and deposition, which is usually followed by glomerular inflammation.
Autoantibodies may be directly involved in the pathogenesis of some diseases, while in others may serve simply as disease markers without a known pathogenic role. For ex-ample, the anti-Sm antibodies found exclusively in patients with SLE are not known to play a pathogenic role. However, in many other situations autoantibodies can trigger various pathogenic mechanisms leading to cell or tissue destruction (Table 16.6).

· Complement-fixing antibodies (IgG and IgM) to red cells may cause intravascular red cell lysis if the complement activation sequence proceeds all the way to the formation of the membrane attack complex or may induce phagocytosis and extravascular lysis if the sequence is stopped at the C3 level, due to the accu-mulation of C3b fragments on the red cell membrane.
· If the antigen-antibody reaction takes place in tissues, pro-inflammatory complement fragments (C3a, C5a) are generated and attract granulocytes and mononuclear cells, which can release proteolytic enzymes and toxic radicals in the area of IC deposition, causing tissue damage.
· Other autoantibodies may have a pathogenic role dependent not on causing cell or tis-sue damage, but on interference with cell functions resulting from their bind-ing to physiologically important cell receptors.
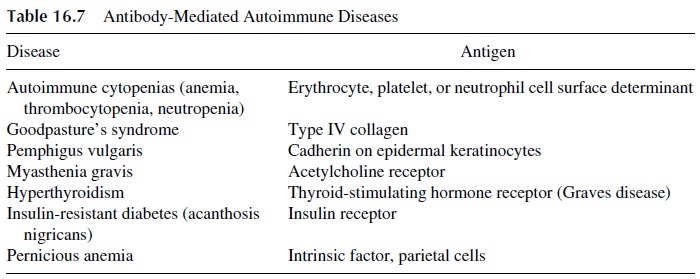
Representative human autoimmune diseases in which auto-antibodies are believed to play a major pathogenic role are listed in Table 16.7. It must be noted in some of these dis-eases there is also a cell-mediated immunity component. For example, in myasthenia gravis, autoreactive T lymphocytes have been described, and both autoreactive cell lines and clones have been successfully established from patient’s lymphocytes. These T lym-phocytes may provide help to autoreactive B lymphocytes producing anti-acetylcholine re-ceptor antibodies. In such cases, autoreactive T lymphocytes could be more central in the pathogenesis of the disease than auto-antibody producing B lymphocytes. However, the pathogenic role of autoantibodies is evident from the fact that newborns to mothers with myasthenia gravis develop myasthenia-like symptoms for as long as they have maternal au-toantibodies in circulation.
2. The Pathogenic Role of Immune Complexes in Autoimmune Diseases
In autoimmune diseases, there is ample opportunity for the formation of immune complexes (IC) involving autoantibodies and self antigens. Several factors determine the pathogenic-ity of IC. They include the size of the IC (inter-mediate-size IC are the most pathogenic), the ability of the host to clear IC (individuals with low complement levels or deficient Fc receptor and/or complement receptor function have delayed IC clearance rates and are prone to develop autoimmune diseases), and physico-chemical properties of IC (which determine the ability to activate complement and/or the deposition in specific tissues). In many occasions, IC are formed in situ, activate the com-plement system, complement split products are formed, and neutrophils are attracted to the area of IC deposition where they will mediate the IC-mediated tissue destruction.
SLE and polyarteritis nodosa are two classical examples of autoimmune diseases in which IC play a major pathogenic role. In SLE, DNA and other nuclear antigens are pre-dominantly involved in the formation of IC, while in polyarteritis nodosa, the most fre-quently identified antigen is hepatitis B surface antigen.
3. The Role of Activated T Lymphocytes in the Pathogenesis of Autoimmune
Typical T-cell–mediated autoimmune diseases are summarized in Table 16.8. T lympho-cytes that are involved in the pathogenesis of such autoimmune diseases may:
· Be autoreactive and recognize self antigens
· Recognize foreign antigen associated with self determinants (modified self)
· Respond to foreign antigens but still induce self tissue destruction.
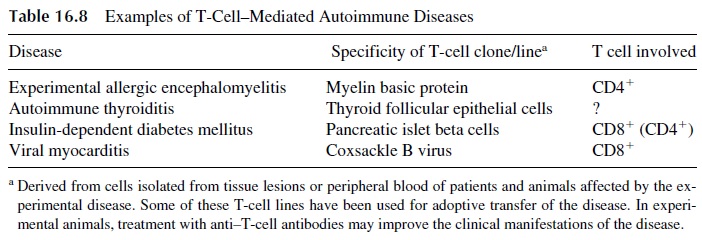
Cytotoxic CD8+ lymphocytes play a pathogenic role in some autoimmune diseases, usually involving the recognition of nonself peptides expressed in the context of self MHC and destroying the cell expressing such “modified” self. For example, coxsackie B virus–defined antigens expressed on the surface of myocardial cells may induce CD8+ -me-diated tissue destruction, causing a virus-induced autoimmune myocarditis.
Activated CD4+ helper cells appear to be frequently involved in cell-mediated au-toimmune reactions. Their pathogenic effects are mediated by the release of cytokines (IFN-γ , IL-1, L-2, IL-4, etc.), which can either trigger inflammatory reactions (if TH1 cells are predominantly involved) or activate autoantibody-producing B lymphocytes (if TH 2 cells are predominantly involved). As a rule, TH1 cells appear to play a dominant role in many organ-specific autoimmune diseases, and TH 2 cells are predominantly involved in systemic autoimmune diseases, such as SLE or rheumatoid arthritis.
D. Pathogenic Factors Involved in the Onset of Autoimmune Diseases
Multiple factors have been proposed as participating in the pathogenesis of autoimmune diseases. These factors can be classified as immunological, genetic, environmental, and hormonal. Each group of factors is believed to contribute in different ways to the patho-genesis of different diseases.
1.Abnormal Immunoregulation
Multiple lymphocyte abnormalities have been described in patients with autoimmune dis-eases. Prominent among them are B-lymphocyte overactivity, presence of spontaneously activated T and B lymphocytes, and decreased suppressor T-cell function.
2. Anti-idiotypic Antibodies
These antibodies may play an important role in autoimmunity. For example, during a nor-mal immune response to a virus, an immune response directed against the viral structures mediating attachment to its target cell is likely to be triggered. As the immune response evolves, anti-idiotypic antibodies reacting with the antigen-binding site of the antiviral an-tibodies may develop. These anti-idiotypes, by recognizing the “internal image of the anti-gen” (which is the configuration of the binding site of the first antibody), may be able to combine with the virus receptor protein in the cell. If the membrane protein used as bind-ing site by the virus happens to be a receptor with important physiological functions, the synthesis of anti-idiotypic antibodies may have adverse effects by either activating or blocking the physiological activation of these functions.
Such a mechanism could explain the origin of antibodies against the acetylcholine and the TSH receptors detected in myasthenia gravis and Grave’s disease, respectively. A hypothetical viral infection would trigger the synthesis of anti-idiotypic antibodies cross-reacting with the acetylcholine receptor at the neuromuscular junction or with the TSH re-ceptor on thyroid cells; these antibodies could interfere with normal function, cause cell death, or induce cell stimulation depending upon their isotype and the receptor epitopes to which they bind.
3. Genetic Factors
Clinical observations have documented increased frequency of autoimmune diseases in families and increased rates of clinical concordance in monozygotic twins. Several studies have also documented associations between HLA antigens and various diseases. As stated earlier, autoimmune diseases can be classified in two groups: one apparently associated with MHC-I genetic markers, and the other associated with MHC-II genetic markers.
The classical example of linkage with MHC-I markers is the association between HLA-B27 and inflammatory spondyloarthropathies (ankylosing spondylitis, Reiter’s syn-drome, etc.). The pathogenic relevance of HLA-B27 is strongly supported by experiments with transgenic mice carrying the gene for HLA-B27. Those transgenic animals sponta-neously develop inflammatory disease involving the gastrointestinal tract, peripheral and vertebral joints, skin, nails, and heart. The disease induced in transgenic mice resembles strikingly the B27-associated disorders that afflict humans with that gene. It has been pos-tulated that the autoimmune reaction is triggered by an infectious peptide presented by HLA-B27 and followed by cross-reactive lymphocyte activation by an endogenous colla-gen-derived peptide, equally associated with HLA-B27.
The linkage of autoimmunity with MHC-II markers is better understood. With the ex-panded definition of MHC-II alleles due to the development of antisera and DNA probes and because of the successful sequencing of the genes coding for the constitutive polypep-tide chains of MHC-II molecules, the significance of MHC-II alelles has become clear. For example, insulin-dependent diabetes mellitus (IDDM) is strongly associated with serolog-ically defined MHC-II markers (HLA-DR3 and HLA-DR4) but is even more strongly cor-related with the presence of uncharged amino acids at position 57 of the chain of DQ (DQB) . Those MHC-II molecules may be critically involved in the pre-sentation of diabetogenic peptides to the immune system.
The exact mechanisms responsible the association between HLA alleles and disease susceptibility are being actively investigated. Two have been hypothesized, based on the persistence and later activation of autoreactive clones:
1. Molecular mimicry, i.e., cross-reactivity between peptides derived from infec-tious agents and peptides derived from autologous proteins which are expressed by most normal resting cells in the organism. Anergic autoreactive T-cell clones would be activated by an immune response against an infectious agent due to this type of cross-reactivity.
2. Lack of expression of MHC alleles able to bind critical endogenous peptides. Under these circumstances potentially autoreactive T-lymphocyte clones would not be eliminated and would remain available for later activation due to an unre-lated immune response or by the presentation of a cross-reactive peptide.
Another important genetic determinant of autoreactivity is the TcR repertoire of a given individual. Several autoimmune diseases show associations with specific TcR vari-able-region types. This is not surprising because the specific recognition of different oligopeptides by different T lymphocytes depends on the diversity of the TcR. Therefore, the development of an autoimmune response should require that the genome of an individual includes genes encoding a particular array of V region genes whose transcription resulted in the expression of TcR able to combine with a specific au-tologous peptide. In addition, the clones expressing such receptors must not be deleted dur-ing embryonic differentiation.
These postulates are supported by immunogenetic studies in different animals and humans with different manifestations of autoimmunity. Those studies suggest that linkages between specific TcR V-region genes and specific autoimmune diseases may actually ex-ist (e.g., IDDM, multiple sclerosis, and SLE). Experimental corollaries of the postulated positive association between specific TcR V-region genes and disease are found in exper-imental allergic encephalomyelitis and murine collagen-induced arthritis. The lymphocytes obtained from arthritic joints of mice susceptible to collagen-induced arthritis have a very limited repertoire of Vβ genes. In mouse strains that do not develop collagen-induced arthritis there are extensive deletion of Vβ genes, including those preferentially expressed by susceptible mice.
Even in identical twins, however, concordance for a particular autoimmune disease never exceeds 40%, suggesting that the presence of autoimmunity-associated TcR V-region genes is not sufficient to cause disease by itself. Indeed, with certain exceptions, hu-man autoimmune diseases are multigenic and the number of the involved genes has not been determined. Recent studies have undertaken genome-wide searches to locate loci and genes that are involved in humans with systemic autoimmune diseases and the corre-sponding animal models. The studies use microsatellite markers and patients from families with multiple afflicted members. Progress has been made, but definitive results are not yet available.
In mice that develop a disease resembling SLE at least 31 different genes contribut-ing to the development of the disease have been identified so far. Study of congenic mice has shed some light into how different loci contribute, through positive and negative epistatic interactions, to loss of tolerance to autoantigens and the progressive establishment of a lupus-like disease. The number of genes involved in human lupus must be significantly higher. A similarly high number of loci have been proposed for diabetes and arthritis in hu-mans and laboratory animals.
4. Environmental Factors
The most important environmental factors are believed to be foreign antigens sharing struc-tural similarity with self-determinants. Exposure to these epitopes can trigger autoimmune reactions. The termmolecular mimicry is used to describe identity or similarity of either amino acid sequences or structural epitopes between foreign and self antigens.
One of the best known examples of autoimmunity resulting from the exposure to cross-reactive antigens is the cardiomyopathy that complicates many cases of acute rheumatic fever. Group A β-hemolytic streptococci have several epitopes cross-reactive with tissue antigens. One of them cross-reacts with an antigen found in cardiac myosin. The normal immune response to such a cross-reactive strain ofStreptococcus will generate lym-phocyte clones that will react with myosin and induce myocardial damage long after the in-fection has been eliminated.
Several other examples of molecular mimicry have been described, as summarized in Table 16.9, and additional ones await better definition. For example, molecular mimicry between the envelope glycolipids of gram-negative bacteria and the myelin of the periph-eral nerves may explain the association of the Gullain-Barré syndrome with Campylobac-
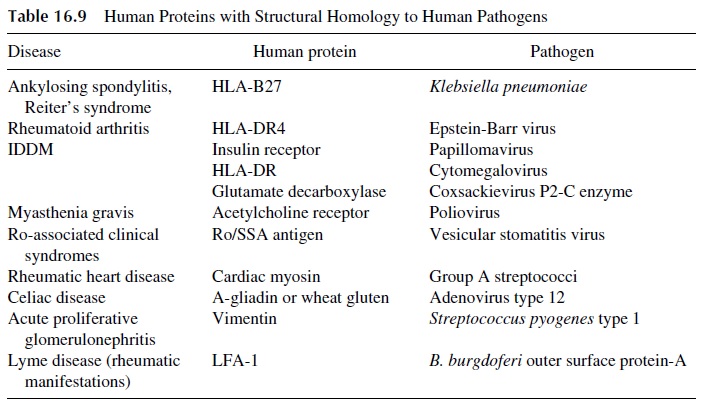
Mimicry between LFA-1 and the Borrelia burgdorferi outer surfaceprotein A is considered responsible for the rheumatic manifestations of Lyme’s disease. Mimicry between glutamate decarboxylase, an enzyme concentrated in pancreatic β cells, and coxsackievirus P2-C, an enzyme involved in the replication of coxsackievirus B, has been considered responsible for the development of insulin-dependent diabetes in humans and in murine models of this disease.
Infectious agents, particularly viruses, can precipitate autoimmunity by inducing the release of sequestered antigens. In autoimmune myocarditis, associated with coxsackie B3 virus, the apparent role of the virus is to cause the release of normally sequestered intra-cellular antigens as a consequence of virus-induced myocardial cell necrosis. Autoanti-bodies and T lymphocytes reactive with sarcolemma and myofibril antigens or peptides de-rived from these antigens emerge, and the autoreactive T lymphocytes are believed to be responsible for the development of persistent myocarditis.
On the other hand, latent viral infections are believed to be responsible for the de-velopment of many autoimmune disorders. Latent infection is commonly associated with integration of the viral genome into the host chromosomes, and while integrated viruses very seldom enter a full replicative cycle and do not cause cytotoxicity, they can inter-fere, directly or indirectly, with several functions of the infected cells. For example, vi-ral proteins may interfere with the function of proteins involved in the control of cell death and survival, such as p53 and bcl-2. In addition, viral proteins may mimic chemokine receptors or ligands and therefore mislead the function of immune cells. T-cell activation secondary to an in apparent viral infection has the potential to in-duce autoimmunity secondary to the release of interferon-γ and TNF, both known to be potent inducers of MHC-II antigen expression. The increased expression of class II MHC antigens would then create optimal conditions for the onset of an autoimmune response directed against MHC-II–self peptide complexes. Such a mechanism has been proposed to explain the onset of autoimmune thyroiditis. An unknown nonlytic virus would cause T-lymphocyte activation in the thyroid gland, followed by increased expression of MHC-II and thyroid-derived peptides, and finally, an antithyroid immune reaction would develop.
Finally, physical trauma can also lead to immune responses to sequestered antigens. The classical example is sympathetic ophthalmia, an inflammatory process of apparent au-toimmune etiology affecting the normal eye after a penetrating injury of the other. This pro-cess may not be limited to trauma; tissue injury induced by any cause may result in the gen-eration of autoreactive T cells that recognize previously cryptic epitopes. Over time, this process will lead to expansion of the immune response by facilitating the onset of immune responses to additional epitopes. This process is known as “epitope spreading.”
5. Central Issues in Molecular Mimicry
There are still difficulties in proving that infections are the cause of autoimmune diseases. The infection may have resolved long before the appearance of disease or the infection may be in apparent, thus obscuring the temporal association between infection and au-toimmunity. Alternatively, molecular mimicry may be the result rather than the cause of autoimmunity. The infectious process may result in the alteration of nonimmunogenic antigenic determinants as a consequence of tissue injury. The autoantibodies that appear when the disease is diagnosed may then be the result of tissue injury rather than the cause of it.
E. Animal Models of Autoimmunity
Our understanding of autoimmune disease has been facilitated by studies in animal mod-els. Several animal models have been developed, each sharing some characteristics of a hu-man disease of autoimmune etiology. These animal models often provide the only experi-mental approaches to the study of the pathogenesis of autoimmune diseases.
In some experimental models, injecting normal animals with antigens extracted from the human target tissues induces autoimmune diseases. A rapid onset and an acute course characterize the resulting diseases. These models have been particularly useful in the study of autoimmune thyroiditis and arthritis (collagen-induced arthritis). Most useful for the study of autoimmunity are animals that develop autoimmune disease spontaneously, whose course is protracted and parallels closely the disease as seen in humans. Representative an-imal models of different autoimmune diseases are listed in Table 16.10.
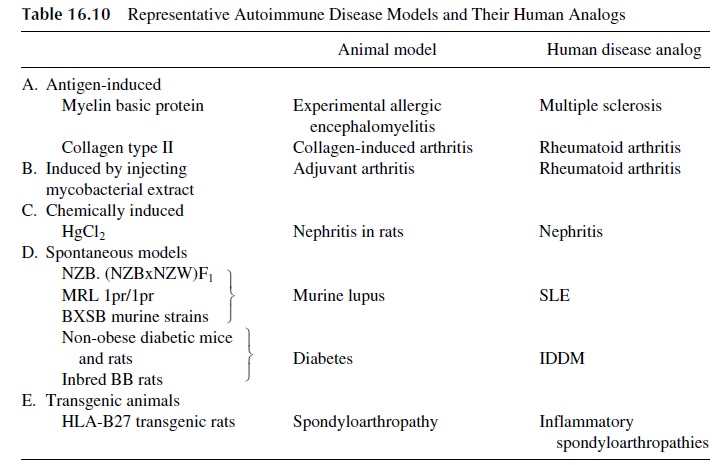
1. Experimental Allergic Encephalomyelitis
Experimental allergic encephalomyelitis (EAE) in mice and rats is the best characterized experimental model of organ-specific autoimmune disease. Immunizing animals with myelin basic protein and adjuvant induces the disease. One to 2 weeks later, the animals develop encephalomyelitis characterized by perivascular mononuclear cell infiltrates and demyelination. The mononuclear cell infiltrates show a predominance of CD4+ helper T lymphocytes, which upon activation release cytokines that attract phagocytic cells to the area of immunological reaction; those cells are, in turn, activated and release enzymes re-sponsible for the demyelination. CD4+ T-lymphocyte clones from animals with EAE dis-ease can transfer the disease to normal animals of the same strain. Genetic manipulations leading to deletion of the genes coding for two specific variable regions of the TcR β chain (Vβ8 and Vβ13) prevent the expression of disease. These two Vβ regions must obviously be involved in the recognition of a dominant epitope of human myelin.
2. Diabetes
Diabetes develops spontaneously in inbred BB rats, as well as in nonobese diabetic (NOD) mice. In both strains the onset of the disease is characterized by T-cell–mediated insulitis, which evolves into diabetes. This disease demonstrates H-2 linkage remarkably similar to that observed between human IDDM and HLA-DR3, DR4, and other MHC-II alleles. In NOD mice, a decreased expression of MHC-I genes, secondary to a TAP-1 gene defi-ciency, has been recently characterized. Such a deficiency would prevent these animals from deleting autoreactive clones during T-cell differentiation.
3. Systemic Lupus Erythematosus
A number of murine strains spontaneously develop autoimmune disease that resembles hu-man SLE:
· (NZB×NZW)F1 female mice develop glomerulonephritis, hemolytic anemia, and anti-DNA antibodies. Numerous alterations in T- and B-lymphocyte function, cytokine release, and macrophage functions have been described in these ani-mals.
· MRL-Ipr/Ipr mice, which lack Fas antigen, and gld mice, which lack Fas ligand, pro-duce autoantibodies and develop arthritis and kidney disease, but they also de-velop massive lymphadenopathy, which is not seen in human disease.
· BXSB mice develop anti-DNA antibodies, nephritis, and vasculitis. In this strain, in contrast to the others, disease susceptibility is linked to the Y chromosome.
Related Topics