Chapter: Pathology: Renal Pathology
Tumors of the Kidney
TUMORS OF THE KIDNEY
Benign tumors of the kidney are as follows:
•
Cortical
adenomas are small, encapsulated cortical nodules measuring <3 cm; they are a common finding at autopsy. They may
be composed of tubular or papillary structures. The papillary adenomas share
the same chromosomal gains as papillary renal cell carcinoma.
•
Angiomyolipomas
are
hamartomas composed of fat, smooth muscle, and blood vessels, common in
patients with tuberous sclerosis.
•
Oncocytomas
are
large, benign tumors that are resected to rule out renal cell carcinoma when they are found incidentally on
imaging studies. They are brown on cut surface and have abundant pink cytoplasm
on microscopy.
Renal cell carcinoma (RCC) is most common age
50–70, with males affected more than females.
Risk factors include
cigarette smoking, chronic analgesic use, asbestos exposure, chronic renal
failure, acquired cystic disease, and von Hippel-Lindau disease (VHL tumor
suppressor gene).
In 10% of cases, the
“classic” triad occurs:
•
Hematuria
•
Palpable mass
•
Flank pain
A variety of paraneoplastic
syndromes from ectopic hormone production can occur:
•
Polycythemia (erythropoietin production)
•
Hypertension (renin production)
•
Cushing syndrome (corticosteroid synthesis)
•
Hypercalcemia (PTH-like hormone)
•
Feminization or masculinization (gonadotropin release)
Renal cell carcinoma may also
cause secondary amyloidosis, a leukemoid reaction, or eosinophilia.
There is a high incidence of
metastasis on initial presentation. The clinical course is unpredictable.
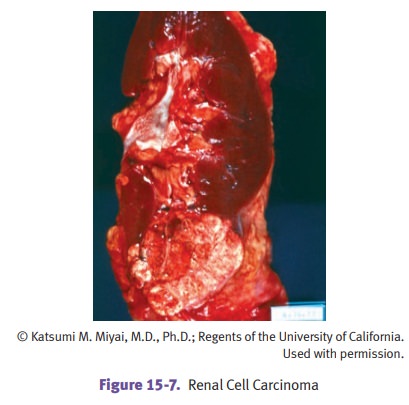
Gross examination typically
demonstrates a large, solitary yellow mass found most commonly in the upper
pole. Areas of necrosis and hemorrhage are commonly present. The tumor often
invades the renal vein and may extend into the inferior vena cava and heart.
Histologic types of RCC are
as follows:
Clear
cell RCC (most common)
•
Often invades renal venous system
•
May have loss of genetic material in 3p
•
A small percent occur in association with von Hippel-Lindau disease
•
Microscopically, there is an alveolar growth pattern with
microcysts
•
Tumor is resistant to chemotherapy and radiotherapy
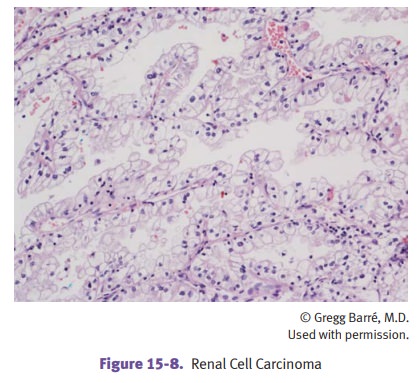
Papillary
RCC
•
Tends to be bilateral and multifocal
•
Cut surface is granular
•
Microscopically, the papillae have a single layer of cells
•
Gains of chromosome 7 and 17 are common
•
Duplications of chromosome 7 increase dosage of protooncogene MET
Chromophobe
RCC (rare)
•
Have cells that stain more darkly than clear cell RCC
•
Have loss of multiple chromosomes
•
Least aggressive of the RCCs
Wilms
tumor (nephroblastoma) typically presents age 2-5 as a large abdominal
mass.
Patients with WAGR, DDS, or
BWS syndrome are at increased risk of Wilms tumor.
•
WAGR syndrome is the cluster of Wilms tumor, aniridia, genital anomalies,
and mental retardation.
•
Beckwith-Wiedemann syndrome (BWS) is an overgrowth disorder with
char-acteristic features and an attendant increased risk of cancer.
•
Denys-Drash syndrome (DDS) affects the genitalia and kidneys.
Both WAGR and DDS are associated
with deletions and mutations, respectively, of the WT1 gene. BWS arises through imprinting abnormalities at the WT2 locus.
Pathologically, Wilms tumor
causes a large, solitary tan mass. Microscopic exami-nation reveals a tumor
containing 3 elements: metanephric blastema, epithelial ele-ments (immature
glomeruli and tubules), and stroma.
Treatment is surgery,
chemotherapy, and radiation, which as a combined therapy yields an excellent
prognosis. Long-term survival rate is 90%.
Transitional cell carcinomas can involve the renal pelvis as well as the
urinary bladder.
Related Topics