Chapter: Pathology: Renal Pathology
Primary Glomerulopathies (Nephritic Syndrome)
PRIMARY GLOMERULOPATHIES (NEPHRITIC SYNDROME)
Acute poststreptococcal glomerulonephritis
(APSGN) (or acute proliferative glomer-ulonephritis or postinfectious
glomerulonephritis) is an immune complex disease that typically occurs 2–4
weeks after a streptococcal infection of the throat or skin. There is a
decreasing incidence in the United States; children are affected more often
than adults.
The infecting organism is
most commonly β -hemolytic group A streptococci,
but APSGN can also be caused by other bacteria, viruses, parasites, and even
systemic diseases (SLE and polyarteritis nodosa). Clinically, it presents with
nephritic syn-drome with elevated antistreptolysin O (ASO) titers (when related
to streptococcal infection) and low C3.
Renal biopsy. Light microscopy shows an infiltrate
of neutrophils in the glomeruli; the process is diffuse, that
is, it involves all the glomeruli. Immunofluorescence shows granular deposits
of IgG and C3 throughout the glomerulus within the cap-illary walls and some
mesangial areas. Electron microscopy shows subepithelial immune complex
deposits (humps).
Treatment is conservative
fluid management. Most children (>95%) have a good prognosis, with complete
recovery, but severe disease can also occur (RPGN 1% and chronic
glomerulonephritis 2%). In adults, 15-50% develop end-stage disease.
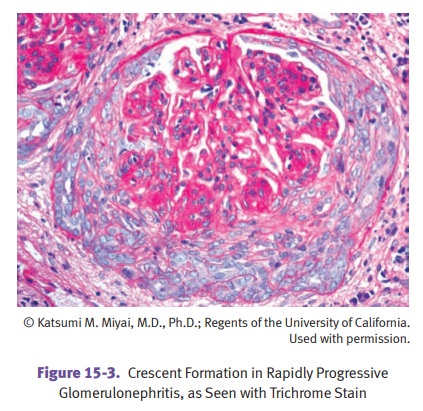
Rapidly progressive glomerulonephritis (RPGN)
(crescentic glomerulonephritis) is group of diseases
characterized by glomerular crescents and a rapid deterioration of renal
function.
Anti-glomerular basement membrane
antibody-mediated crescentic glomer ulonephritis has a peak incidence at age
20-40, and males are affected more frequently than females.
Pulmonary involvement is
called Goodpasture syndrome. These
patients have pul-monary hemorrhage and hemoptysis.
Renal biopsy findings include
hypercellularity, crescents, and fibrin deposition in glomeruli.
Immunofluorescence shows a smooth and linear pattern of IgG and C3 in the
glomerular basement membrane (GBM). By electron microscopy, there are no
deposits, but there is glomerular basement membrane disruption.
Even with treatment (plasma
exchange, steroids, and cytotoxic drugs), the prognosis is poor because of
risks of severe and life-threatening pulmonary hemorrhage and renal failure.
Early aggressive treatment may prevent end-stage renal failure.
Immune-complex mediated crescentic
glomerulonephritis
•
Any of the immune complex nephritides can cause crescent formation.
•
IF shows a granular pattern and EM shows discrete deposits.
Pauci-immune crescentic glomerulonephritis
•
Antineutrophilic cytoplasmic antibodies (ANCA) are present in
serum.
•
IF and EM are negative for immunoglobulins and complement
components.
IgA nephropathy (Berger disease) is the most
common cause of glomerulonephritis in the world, being
particularly common in France, Japan, Italy, and Austria. It affects children
and young adults (mostly males).
IgA nephropathy is
characterized by recurrent gross hematuria (a predominately nephritic
presentation), whose onset may follow a respiratory infection. IgA nephropathy
can be associated with celiac sprue and Henoch-Schönlein purpura or can be
secondary to celiac sprue or liver disease.
The pathogenesis is unknown,
but may be related to a possible entrapment of cir-culating immune complexes
with activation of the alternate complement pathway; it may also be related to
a genetic predisposition.
Renal biopsy. Light microscopy may show
normal glomeruli or mesangial prolif-eration. Immunofluorescence shows
mesangial deposits of IgA and C3. Electron microscopy shows mesangial immune
complex deposits.
Many cases slowly progress to
renal failure over 25 years.
Membranoproliferative glomerulonephritis (MPGN) is a form of
glomerular disease that affects both the
glomerular mesangium and the basement membranes; it occurs in 2 types, Type I
and Type II (dense deposit disease).
The clinical presentation is
variable, and may be nephritic, nephrotic, or mixed. MPGN may be secondary to
many systemic disorders (systemic lupus erythemato-sus, endocarditis), chronic
infections (HBV, HCV, HIV), and malignancies (chronic lymphocytic leukemia).
Laboratory studies show
decreased serum C3 and the presence of C3 nephritic fac-tor (MPGN type II).
Light microscopy demonstrates
a lobulated appearance of the glomeruli due to mesangial and endothelial cell
proliferation and/or deposition of subendothelial immune complex deposits.
Splitting of the basement membrane (“tram-tracking”) may be seen with a silver or
periodic acid-Schiff (PAS) stain.
Immunofluorescence in type I MPGN shows a granular pattern of C3
often with IgG, C1q, and C4; in type
II, immunofluorescence shows a granular and linear pat-tern of C3 deposition.
Electron microscopy in type I shows subendothelial and mesangial
immune com-plex deposits, and in type II shows dense deposits within the
glomerular basement membrane.
MPGN typically has a slowly
progressive course, resulting in chronic renal failure over the course of 10
years. There is a high incidence of recurrence in transplants.
Alport syndrome is a rare X-linked disorder
caused by a defect in type IV collagen. It is characterized by
hereditary nephritis, hearing loss, and ocular abnormalities. The most common
mutation causing Alport is in the COL4A5
gene coding for the alpha-3, -4, and -5 chain of type IV collagen.
Gross or microscopic
hematuria begins in childhood. Hearing loss (leading to sen-sorineural
deafness) and various ocular abnormalities of the lens and cornea can occur.
Alport is a progressive disease that ultimately results in renal failure.
Electron microscopy shows
alternating thickening and thinning of basement mem-brane with splitting of the
lamina densa, causing a “basketweave” appearance.
Related Topics