Chapter: Basic & Clinical Pharmacology : Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action
The Time Course of Drug Effect
THE TIME COURSE OF DRUG EFFECT
The
principles of pharmacokinetics and those of pharmacodynamics provide a
framework for understanding the time course of drug effect.
Immediate Effects
In
the simplest case, drug effects are directly related to plasma concentrations,
but this does not necessarily mean that effects simply parallel the time course
of concentrations. Because the relationship between drug concentration and
effect is not linear (recall the Emax model described), the effect
will not usually be linearly proportional to the concentration.
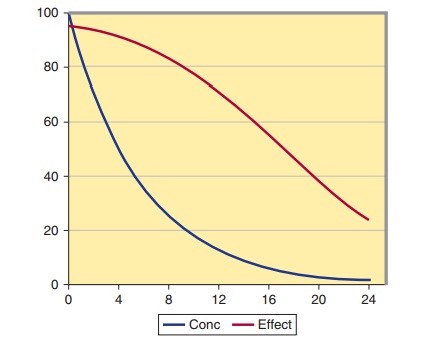
Consider
the effect of an angiotensin-converting enzyme (ACE) inhibitor, such as
enalapril, on plasma ACE. The half-life of enalapril is about 3 hours. After an
oral dose of 10 mg, the peak plasma concentration at 3 hours is about 64 ng/mL.
Enalapril is usually given once a day, so seven half-lives will elapse from the
time of peak concentration to the end of the dosing interval. The concentration
of enalapril after each half-life and the correspond-ing extent of ACE
inhibition are shown in Figure 3–5. The extent of inhibition of ACE is
calculated using the Emax model, where Emax, the maximum
extent of inhibition, is 100% and the C50, the concentration of the
drug that produces 50% of maximum effect, is about 1 ng/mL.
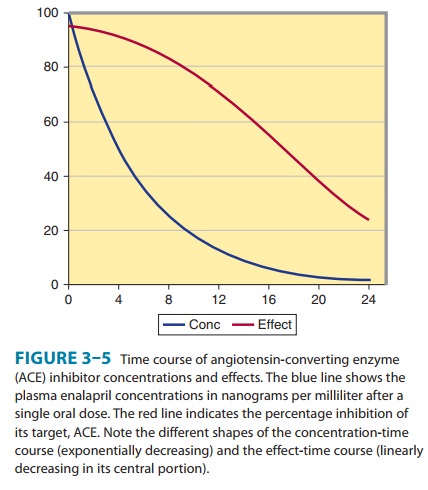
Note that plasma concentrations of enalapril change by a factor of 16 over the first 12 hours (four half-lives) after the peak, but ACE inhibition has only decreased by 20%. Because the concen-trations over this time are so high in relation to the C50, the effect on ACE is almost constant. After 24 hours, ACE is still 33% inhibited. This explains why a drug with a short half-life can be given once a day and still maintain its effect throughout the day. The key factor is a high initial concentration in relation to the C50. Even though the plasma concentration at 24 hours is less than 1% of its peak, this low concentration is still half the C50. This is very common for drugs that act on enzymes (eg, ACE inhibitors) or compete at receptors (eg, propranolol).
When
concentrations are in the range between four times and one fourth of the C50,
the time course of effect is essentially a linear function of time. It takes
four half-lives for concentrationsto drop from an effect of 80% to 20% of Emax—15%
of the effect is lost every half-life over this concentration range. At
concentra-tions below one fourth the C50, the effect becomes almost
directly proportional to concentration and the time course of drug effect will
follow the exponential decline of concentration. It is only when the
concentration is low in relation to the C 50 that the con-cept of a
“half-life of drug effect” has any meaning.
Delayed Effects
Changes
in drug effects are often delayed in relation to changes in plasma
concentration. This delay may reflect the time required for the drug to
distribute from plasma to the site of action. This will be the case for almost
all drugs. The delay due to distribution is apharmacokinetic phenomenon that
can account for delays of a few minutes. This distributional delay can account
for the lag of effects after rapid intravenous injection of central nervous
system (CNS)–active agents such as thiopental.
A
common reason for more delayed drug effects—especially those that take many
hours or even days to occur—is the slow turnover of a physiologic substance
that is involved in the expression of the drug effect. For example, warfarin
works as an anticoagulant by inhibiting vitamin K epoxidase in the liver. This
action of warfarin occurs rap-idly, and inhibition of the enzyme is closely
related to plasma concen-trations of warfarin. The clinical effect of warfarin, eg, on the International Normalized
Ratio (INR), reflects a decrease in the con-centration of the prothrombin
complex of clotting factors. Inhibition of vitamin K epoxidase decreases the
synthesis of these clotting fac-tors, but the complex has a long half-life
(about 14 hours), and it is this half-life that determines how long it takes
for the concentration of clotting factors to reach a new steady state and for a
drug effect to reflect the average warfarin plasma concentration.
Cumulative Effects
Some
drug effects are more obviously related to a cumulative action than to a
rapidly reversible one. The renal toxicity of aminoglycoside antibiotics (eg,
gentamicin) is greater when administered as a constant infusion than with
intermittent dosing. It is the accumulation of aminoglycoside in the renal
cortex that is thought to cause renal damage. Even though both dosing schemes
produce the same average steady-state concentration, the intermit-tent dosing
scheme produces much higher peak concentrations, which saturate an uptake
mechanism into the cortex; thus, total aminoglycoside accumulation is less. The
difference in toxicity is a predictable consequence of the different patterns
of concentra-tion and the saturable uptake mechanism.
The
effect of many drugs used to treat cancer also reflects a cumulative action—eg,
the extent of binding of a drug to DNA is proportional to drug concentration
and is usually irreversible. The effect on tumor growth is therefore a
consequence of cumulative exposure to the drug. Measures of cumulative
exposure, such as AUC, provide a means to individualize treatment.
Related Topics