Chapter: Basic & Clinical Pharmacology : Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action
Target Concentration Intervention: Application of Pharmacokinetics & Pharmacodynamics to Dose Individualization
TARGET CONCENTRATION INTERVENTION:
APPLICATION OF PHARMACOKINETICS & PHARMACODYNAMICS TO DOSE
INDIVIDUALIZATION
The
basic principles outlined above can be applied to the interpre-tation of
clinical drug concentration measurements on the basis of three major
pharmacokinetic variables: absorption, clearance, and volume of distribution
(and the derived variable, half-life). In addi-tion, it may be necessary to
consider two pharmacodynamic vari-ables: maximum effect attainable in the
target tissue and the sensitivity of the tissue to the drug. Diseases may
modify all of these parameters, and the ability to predict the effect of
disease states on pharmacokinetic parameters is important in properly adjusting
dosage in such cases. (See Box: The Target Concentration Strategy.)
Pharmacokinetic Variables
A. Absorption
The
amount of drug that enters the body depends on the patient’s adherence to the
prescribed regimen and on the rate and extent of transfer from the site of
administration to the blood.
The Target Concentration Strategy
Recognition of the essential role of concentration in
linking pharmacokinetics and pharmacodynamics leads naturally to the target
concentration strategy. Pharmacodynamic princi-ples can be used to predict the
concentration required to achieve a particular degree of therapeutic effect.
This target concentration can then be achieved by using pharmacoki-netic
principles to arrive at a suitable dosing regimen (Holford, 1999). The target
concentration strategy is a process for opti-mizing the dose in an individual
on the basis of a measured surrogate response such as drug concentration:
Choose the target concentration, TC.
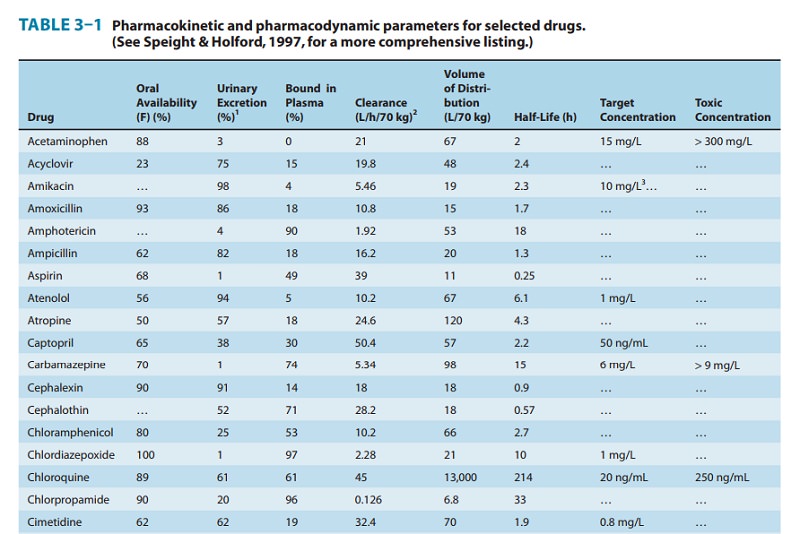
Predict volume of distribution (V) and clearance (CL)
based on standard population values (eg, Table 3–1) with adjustments for
factors such as weight and renal function.
Give a loading dose or maintenance dose calculated
from TC, V, and CL.
Measure the patient’s response and drug
concentra-tion.
Revise V and/or CL based on the measured
concentra-tion.
Repeat steps 3–5, adjusting the predicted dose to
achieve TC.
Overdosage
and underdosage relative to the prescribed dosage—both aspects of failure of
adherence—can frequently be detected by concentration measurements when gross
deviations from expected values are obtained. If adherence is found to be
adequate, absorption abnormalities in the small bowel may be the cause of
abnormally low concentrations. Variations in the extent of bioavailability are
rarely caused by irregularities in the manufac-ture of the particular drug
formulation. More commonly, varia-tions in bioavailability are due to
metabolism during absorption.
B. Clearance
Abnormal
clearance may be anticipated when there is major impairment of the function of
the kidney, liver, or heart. Creatinine clearance is a useful quantitative
indicator of renal function. Conversely, drug clearance may be a useful
indicator of the func-tional consequences of heart, kidney, or liver failure,
often with greater precision than clinical findings or other laboratory tests.
For example, when renal function is changing rapidly, estimation of the
clearance of aminoglycoside antibiotics may be a more accurate indicator of
glomerular filtration than serum creatinine.
Hepatic
disease has been shown to reduce the clearance and prolong the half-life of
many drugs. However, for many other drugs known to be eliminated by hepatic
processes, no changes in clearance or half-life have been noted with similar
hepatic disease. This reflects the fact that hepatic disease does not always
affect the hepatic intrinsic clearance. At present, there is no reliable
markerof hepatic drug-metabolizing function that can be used to predict changes
in liver clearance in a manner analogous to the use of creatinine clearance as
a marker of renal drug clearance.
C. Volume of Distribution
The
apparent volume of distribution reflects a balance between binding to tissues,
which decreases plasma concentration and makes the apparent volume larger, and
binding to plasma pro-teins, which increases plasma concentration and makes the
appar-ent volume smaller. Changes in either tissue or plasma binding can change
the apparent volume of distribution determined from plasma concentration
measurements. Older people have a relative decrease in skeletal muscle mass and
tend to have a smaller appar-ent volume of distribution of digoxin (which binds
to muscle proteins). The volume of distribution may be overestimated in obese
patients if based on body weight and the drug does not enter fatty tissues
well, as is the case with digoxin. In contrast, theophyl-line has a volume of
distribution similar to that of total body water. Adipose tissue has almost as
much water in it as other tis-sues, so that the apparent total volume of
distribution of theophyl-line is proportional to body weight even in obese
patients.
Abnormal
accumulation of fluid—edema, ascites, pleural effusion—can markedly increase
the volume of distribution of drugs such as gentamicin that are hydrophilic and
have small vol-umes of distribution.
D. Half-Life
The
differences between clearance and half-life are important in defining the
underlying mechanisms for the effect of a disease state on drug disposition.
For example, the half-life of diazepam increases with patient age. When
clearance is related to age, it is found that clearance of this drug does not
change with age. The increasing half-life for diazepam actually results from
changes in the volume of distribution with age; the metabolic processes
responsible for eliminating the drug are fairly constant.
Pharmacodynamic Variables
A. Maximum Effect
All
pharmacologic responses must have a maximum effect (Emax). No matter
how high the drug concentration goes, a point will be reached beyond which no
further increment in response is achieved.
If
increasing the dose in a particular patient does not lead to a further clinical
response, it is possible that the maximum effect has been reached. Recognition
of maximum effect is helpful in avoid-ing ineffectual increases of dose with
the attendant risk of toxicity.
B. Sensitivity
The
sensitivity of the target organ to drug concentration is reflected by the
concentration required to produce 50% of maxi-mum effect, the C 50.
Diminished sensitivity to the drug can be detected by measuring drug
concentrations that are usually asso-ciated with therapeutic response in a
patient who has not responded. This may be a result of abnormal physiology—eg,
hyperkalemia diminishes responsiveness to digoxin—or drug antagonism—eg,
calcium channel blockers impair the inotropic response to digoxin.
Increased
sensitivity to a drug is usually signaled by exaggerated responses to small or
moderate doses. The pharmacodynamic nature of this sensitivity can be confirmed
by measuring drug concentrations that are low in relation to the observed
effect.
Related Topics