Chapter: Basic & Clinical Pharmacology : Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action
Pharmacokinetics
PHARMACOKINETICS
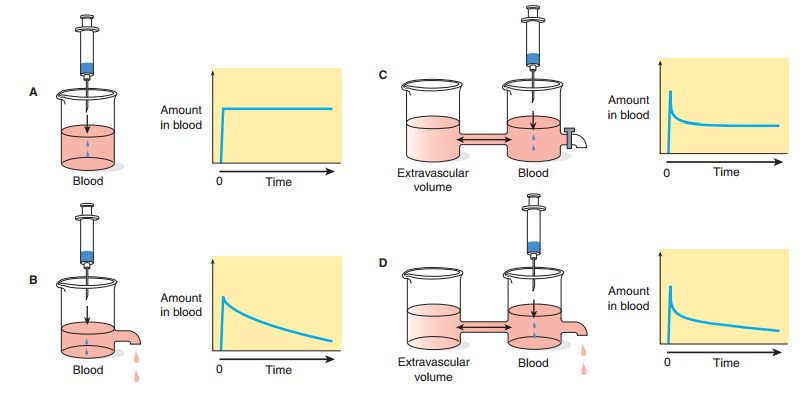
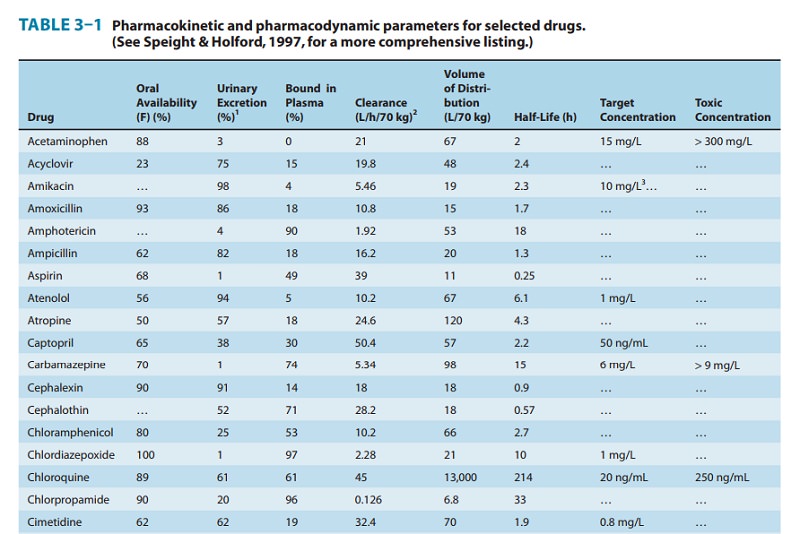
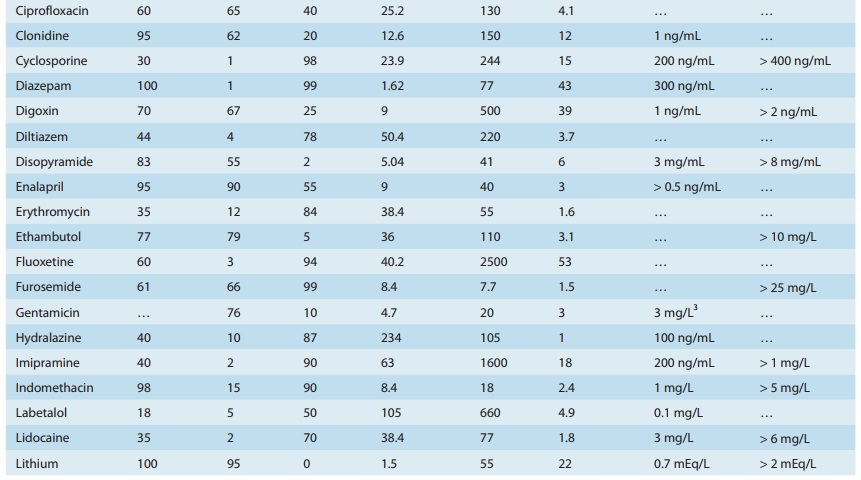
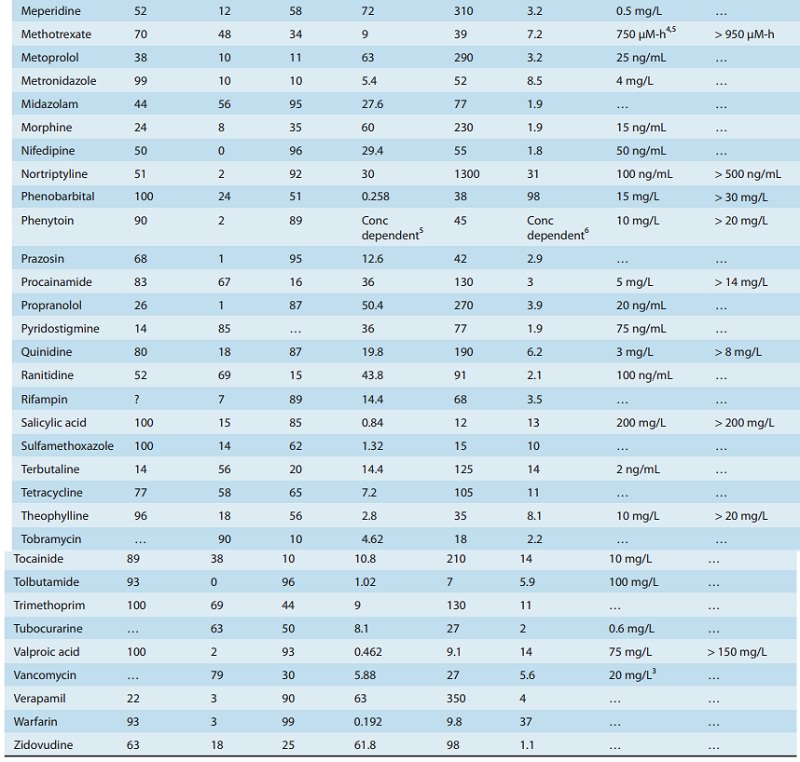
The “standard” dose of a drug is based on trials in healthy volunteers and patients with average ability to absorb, distribute, and eliminate the drug (see Clinical Trials: The IND and NDA). This dose will not be suitable for every patient. Several physiologic pro-cesses (eg, maturation of organ function in infants) and pathologic processes (eg, heart failure, renal failure) dictate dosage adjustment in individual patients. These processes modify specific pharmacokinetic parameters. The two basic parameters are clearance, the measure of the ability of the body to eliminate the drug; and volume of distri-bution, the measure of the apparent space in the body available tocontain the drug. These parameters are illustrated schematically in Figure 3–2 where the volume of the beakers into which the drugs diffuse represents the volume of distribution and the size of the out-flow “drain” in Figures 3–2B and 3–2D represents the clearance.
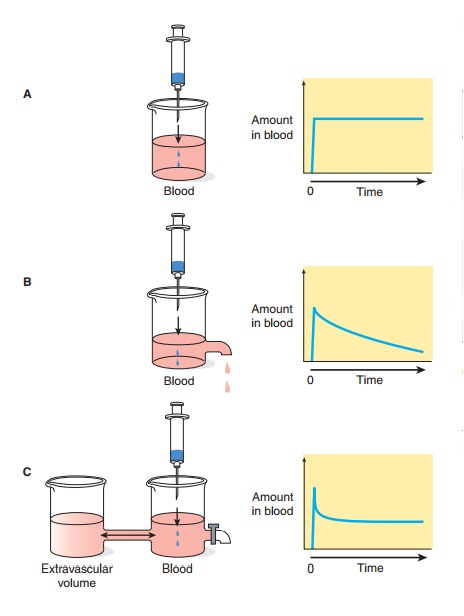
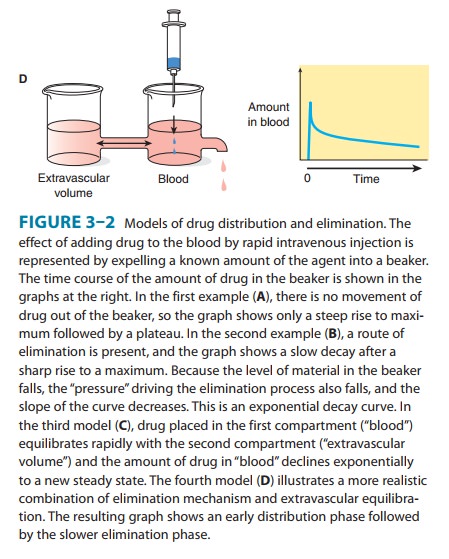
Volume of Distribution
Volume of distribution (V) relates the amount of drug in the body to the concentration of drug (C) in blood or plasma:

The volume of distribution may be defined with respect to blood, plasma, or water (unbound drug), depending on the con-centration used in equation (1) (C = Cb, Cp, or Cu).
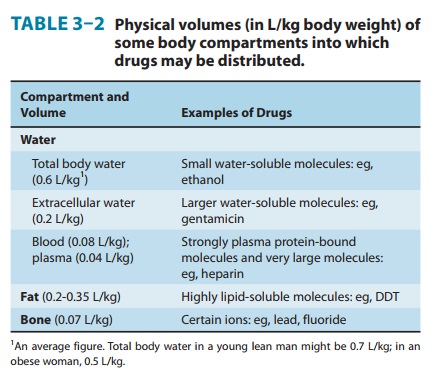
That the V calculated from equation (1) is an apparent volume may be appreciated by comparing the volumes of distribution of drugs such as digoxin or chloroquine (Table 3–1) with some of the physical volumes of the body (Table 3–2). Volume of distri-bution can vastly exceed any physical volume in the body because it is the volume apparently necessary to contain the amount of drug homogeneously at the concentration found in the blood, plasma, or water. Drugs with very high volumes of distribution have much higher concentrations in extravascular tissue than in the vascular compartment, ie, they are not homogeneously distrib-uted. Drugs that are completely retained within the vascular com-partment, on the other hand, have a minimum possible volume of distribution equal to the blood component in which they are dis-tributed, eg, 0.04 L/kg body weight or 2.8 L/70 kg (Table 3–2) for a drug that is restricted to the plasma compartment.
Clearance
Drug clearance principles are similar to the clearance concepts of renal physiology. Clearance of a drug is the factor that predicts the rate of elimination in relation to the drug concentration:

Clearance, like volume of distribution, may be defined with respect to blood (CLb), plasma (CLp), or unbound in water (CLu), depending on the concentration measured.
It is important to note the additive character of clearance. Elimination of drug from the body may involve processes occur-ring in the kidney, the lung, the liver, and other organs. Dividing the rate of elimination at each organ by the concentration of drug presented to it yields the respective clearance at that organ.
Added together, these separate clearances equal total systemic clearance:
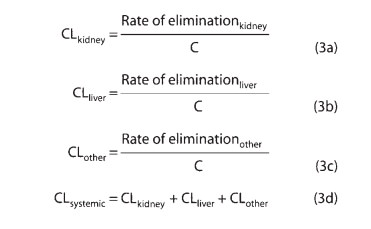
“Other” tissues of elimination could include the lungs and additional sites of metabolism, eg, blood or muscle.The two major sites of drug elimination are the kidneys and the liver. Clearance of unchanged drug in the urine represents renal clear-ance. Within the liver, drug elimination occurs via biotransformation of parent drug to one or more metabolites, or excretion of unchanged drug into the bile, or both. For most drugs, clearance is constant over the concentration range encountered in clinical settings, ie, elimination is not saturable, and the rate of drug elimination is directly proportional to concentration (rearranging equation [2]):
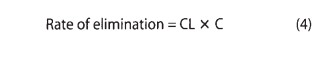
This is usually referred to as first-order elimination. When clearance is first-order, it can be estimated by calculating the areaunder the curve (AUC) of the time-concentration profile after adose. Clearance is calculated from the dose divided by the AUC.
A. Capacity-Limited Elimination
For drugs that exhibit capacity-limited elimination (eg, pheny-toin, ethanol), clearance will vary depending on the concentration of drug that is achieved (Table 3–1). Capacity-limited elimination is also known as mixed-order, saturable, dose- or concentration-dependent, nonlinear, and Michaelis-Menten elimination.
Most drug elimination pathways will become saturated if the dose and therefore the concentration are high enough. When blood flow to an organ does not limit elimination , the relation between elimination rate and concentration (C) is expressed mathematically in equation (5):
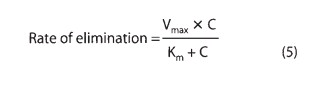
The maximum elimination capacity is Vmax, and Km is the drug concentration at which the rate of elimination is 50% of Vmax. At concentrations that are high relative to the Km, the elimination rate is almost independent of concentration—a state of “pseudo-zero order” elimination. If dosing rate exceeds elimination capac-ity, steady state cannot be achieved: The concentration will keep on rising as long as dosing continues. This pattern of capacity-limited elimination is important for three drugs in common use: ethanol, phenytoin, and aspirin. Clearance has no real meaning for drugs with capacity-limited elimination, and AUC should not be used to describe the elimination of such drugs.
B. Flow-Dependent Elimination
In contrast to capacity-limited drug elimination, some drugs are cleared very readily by the organ of elimination, so that at any clinically realistic concentration of the drug, most of the drug in the blood perfusing the organ is eliminated on the first pass of the drug through it. The elimination of these drugs will thus depend primarily on the rate of drug delivery to the organ of elimination. Such drugs (see Table 4–7) can be called “high-extraction” drugs since they are almost completely extracted from the blood by the organ. Blood flow to the organ is the main determinant of drug delivery, but plasma protein binding and blood cell partitioning may also be important for extensively bound drugs that are highly extracted.
Half-Life
Half-life (t1/2) is the time required to change the amount of drug in the body by one-half during elimination (or during a constant infusion). In the simplest case—and the most useful in designing drug dosage regimens—the body may be considered as a single compartment (as illustrated in Figure 3–2B) of a size equal to the volume of distribution (V). The time course of drug in the body will depend on both the volume of distribution and the clearance:
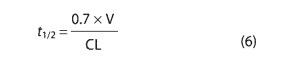
Because drug elimination can be described by an exponential process, the time taken for a twofold decrease can be shown to be proportional to the natural logarithm of 2. The constant 0.7 in equation (6) is an approximation to the natural logarithm of 2.
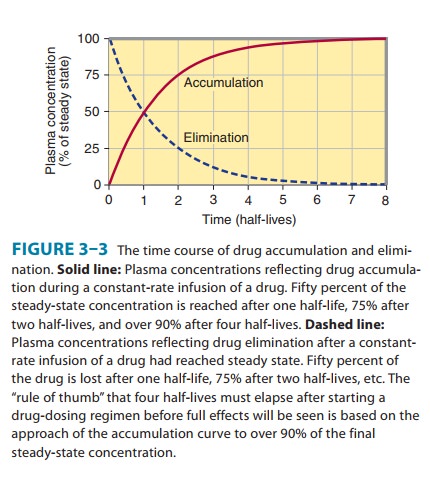
Half-life is useful because it indicates the time required to attain 50% of steady state—or to decay 50% from steady-state conditions—after a change in the rate of drug administration. Figure 3–3 shows the time course of drug accumulation during a constant-rate drug infusion and the time course of drug elimina-tion after stopping an infusion that has reached steady state.
Disease states can affect both of the physiologically related primary pharmacokinetic parameters: volume of distribution and clearance. A change in half-life will not necessarily reflect a change in drug elimination. For example, patients with chronic renal failure have decreased renal clearance of digoxin but also a decreased volume of distribution; the increase in digoxin half-life is not as great as might be expected based on the change in renal function. The decrease in volume of distribution is due to the decreased renal and skeletal muscle mass and consequent decreased tissue binding of digoxin to Na+/K+-ATPase.
Many drugs will exhibit multicompartment pharmacokinetics (as illustrated in Figures 3–2C and 3–2D). Under these condi-tions, the “true” terminal half-life, as given in Table 3–1, will be greater than that calculated from equation (6)
Drug Accumulation
Whenever drug doses are repeated, the drug will accumulate in the body until dosing stops. This is because it takes an infinite time (in theory) to eliminate all of a given dose. In practical terms, this means that if the dosing interval is shorter than four half-lives, accumulation will be detectable.
Accumulation is inversely proportional to the fraction of the dose lost in each dosing interval. The fraction lost is 1 minus the fraction remaining just before the next dose. The fraction remaining can be predicted from the dosing interval and the half-life. A convenient index of accumulation is the accumula-tion factor:
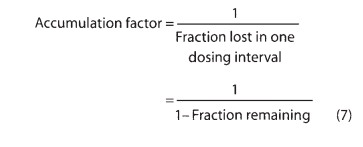
For a drug given once every half-life, the accumulation factor is 1/0.5, or 2. The accumulation factor predicts the ratio of the steady-state concentration to that seen at the same time following the first dose. Thus, the peak concentrations after intermittent doses at steady state will be equal to the peak concentration after the first dose multiplied by the accumulation factor.
Bioavailability
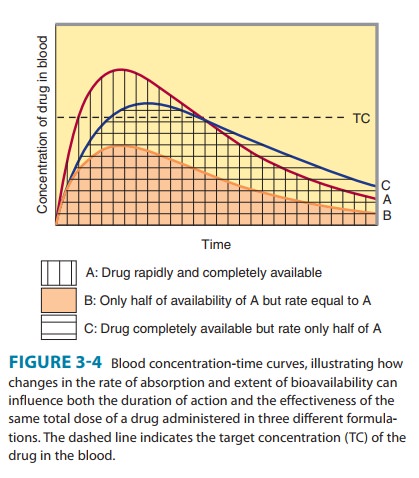
Bioavailability is defined as the fraction of unchanged drug reach-ing the systemic circulation following administration by any route (Table 3–3). The area under the blood concentration-time curve (AUC) is proportional to the extent of bioavailability for a drug if its elimination is first-order (Figure 3–4). For an intravenous dose of the drug, bioavailability is assumed to be equal to unity. For a drug administered orally, bioavailability may be less than 100% for two main reasons—incomplete extent of absorption across the gut wall and first-pass elimination by the liver .
A. Extent of Absorption
After oral administration, a drug may be incompletely absorbed, eg, only 70% of a dose of digoxin reaches the systemic circulation. This is mainly due to lack of absorption from the gut. Other drugs are either too hydrophilic (eg, atenolol) or too lipophilic (eg, acy clovir) to be absorbed easily, and their low bioavailability is also due to incomplete absorption. If too hydrophilic, the drug cannot cross the lipid cell membrane; if too lipophilic, the drug is not soluble enough to cross the water layer adjacent to the cell. Drugs may not be absorbed because of a reverse transporter associated with P-glycoprotein. This process actively pumps drug out of gut wall cells back into the gut lumen. Inhibition of P-glycoprotein and gut wall metabolism, eg, by grapefruit juice, may be associated with substantially increased drug absorption.
B. First-Pass Elimination
Following absorption across the gut wall, the portal blood delivers the drug to the liver prior to entry into the systemic circulation. A drug can be metabolized in the gut wall (eg, by the CYP3A4 enzyme system) or even in the portal blood, but most commonly it is the liver that is responsible for metabolism before the drug reaches the systemic circulation. In addition, the liver can excrete the drug into the bile. Any of these sites can contribute to this reduction in bioavailability, and the overall process is known as first-pass elimination. The effect of first-pass hepatic elimination on bioavailability is expressed as the extraction ratio (ER):

where Q is hepatic blood flow, normally about 90 L/h in a person weighing 70 kg.The systemic bioavailability of the drug (F) can be predicted from the extent of absorption (f ) and the extraction ratio (ER):

A drug such as morphine is almost completely absorbed (f = 1), so that loss in the gut is negligible. However, the hepatic extrac-tion ratio for morphine is morphine clearance (60 L/h/70 kg) divided by hepatic blood flow (90 L/h/70 kg) or 0.67, so (1 – ER) is 0.33. The bioavailability of morphine is therefore expected to be about 33%, which is close to the observed value (Table 3–1).
C. Rate of Absorption
The distinction between rate and extent of absorption is shown in Figure 3–4. The rate of absorption is determined by the site of administration and the drug formulation. Both the rate of absorption and the extent of input can influence the clinical effectiveness of a drug. For the three different dosage forms depicted in Figure 3–4, there would be significant differences in the inten-sity of clinical effect. Dosage form B would require twice the dose to attain blood concentrations equivalent to those of dosage form A. Differences in rate of absorption may become important for drugs given as a single dose, such as a hypnotic used to induce sleep. In this case, drug from dosage form A would reach its target concentration earlier than drug from dosage form C; concentra-tions from A would also reach a higher level and remain above the target concentration for a longer period. In a multiple dosing regi-men, dosage forms A and C would yield the same average blood level concentrations, although dosage form A would show some-what greater maximum and lower minimum concentrations.
The mechanism of drug absorption is said to be zero-order when the rate is independent of the amount of drug remaining in the gut, eg, when it is determined by the rate of gastric emptying or by a controlled-release drug formulation. In contrast, when the dose is dissolved in gastrointestinal fluids, the rate of absorption is usually proportional to the gastrointestinal concentration and is said to be first-order.
Extraction Ratio & the First-Pass Effect
Systemic clearance is not affected by bioavailability. However, clearance can markedly affect the extent of availability because it determines the extraction ratio (equation [8a]). Of course, thera-peutic blood concentrations may still be reached by the oral route of administration if larger doses are given. However, in this case, the concentrations of the drug metabolites will be increased sig-nificantly over those that would occur following intravenous administration. Lidocaine and verapamil are both used to treat cardiac arrhythmias and have bioavailability less than 40%, but lidocaine is never given orally because its metabolites are believed to contribute to central nervous system toxicity. Other drugs that are highly extracted by the liver include isoniazid, morphine, pro-pranolol, and several tricyclic antidepressants (Table 3–1).
Drugs with high extraction ratios will show marked variations in bioavailability between subjects because of differences in hepatic function and blood flow. These differences can explain the marked variation in drug concentrations that occurs among indi-viduals given similar doses of highly extracted drugs. For drugs that are highly extracted by the liver, bypassing hepatic sites of elimination (eg, in hepatic cirrhosis with portosystemic shunting) will result in substantial increases in drug availability, whereas for
drugs that are poorly extracted by the liver (for which the differ-ence between entering and exiting drug concentration is small), shunting of blood past the liver will cause little change in avail-ability. Drugs in Table 3–1 that are poorly extracted by the liver include chlorpropamide, diazepam, phenytoin, theophylline, tolbutamide, and warfarin.
Alternative Routes of Administration & the First-Pass Effect
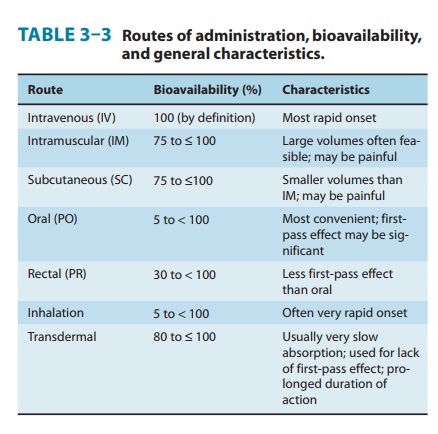
There are several reasons for different routes of administration used in clinical medicine (Table 3–3)—for convenience (eg, oral), to maximize concentration at the site of action and minimize it elsewhere (eg, topical), to prolong the duration of drug absorption (eg, transdermal), or to avoid the first-pass effect.
The hepatic first-pass effect can be avoided to a great extent by use of sublingual tablets and transdermal preparations and to a lesser extent by use of rectal suppositories. Sublingual absorption provides direct access to systemic—not portal—veins. The trans-dermal route offers the same advantage. Drugs absorbed from suppositories in the lower rectum enter vessels that drain into the inferior vena cava, thus bypassing the liver. However, suppositories tend to move upward in the rectum into a region where veins that lead to the liver predominate. Thus, only about 50% of a rectal dose can be assumed to bypass the liver.
Although drugs administered by inhalation bypass the hepatic first-pass effect, the lung may also serve as a site of first-pass loss by excretion and possibly metabolism for drugs administered by nongastrointestinal (“parenteral”) routes.
Related Topics