Chapter: Basic & Clinical Pharmacology : Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action
Clearance - Pharmacokinetics
Clearance
Drug clearance principles are similar to the clearance concepts of renal physiology. Clearance of a drug is the factor that predicts the rate of elimination in relation to the drug concentration:

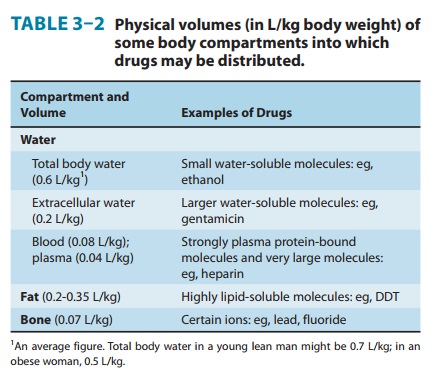
Clearance, like volume of distribution, may be defined with respect to blood (CLb), plasma (CLp), or unbound in water (CLu), depending on the concentration measured.
It is important to note the additive character of clearance. Elimination of drug from the body may involve processes occur-ring in the kidney, the lung, the liver, and other organs. Dividing the rate of elimination at each organ by the concentration of drug presented to it yields the respective clearance at that organ.
Added together, these separate clearances equal total systemic clearance:
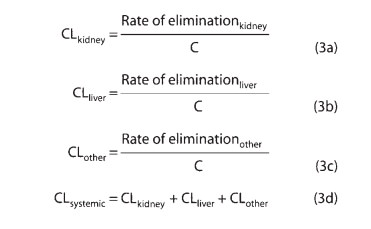
“Other” tissues of elimination could include the lungs and additional sites of metabolism, eg, blood or muscle.The two major sites of drug elimination are the kidneys and the liver. Clearance of unchanged drug in the urine represents renal clear-ance. Within the liver, drug elimination occurs via biotransformation of parent drug to one or more metabolites, or excretion of unchanged drug into the bile, or both. For most drugs, clearance is constant over the concentration range encountered in clinical settings, ie, elimination is not saturable, and the rate of drug elimination is directly proportional to concentration (rearranging equation [2]):
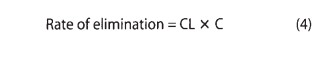
This is usually referred to as first-order elimination. When clearance is first-order, it can be estimated by calculating the areaunder the curve (AUC) of the time-concentration profile after adose. Clearance is calculated from the dose divided by the AUC.
A. Capacity-Limited Elimination
For drugs that exhibit capacity-limited elimination (eg, pheny-toin, ethanol), clearance will vary depending on the concentration of drug that is achieved (Table 3–1). Capacity-limited elimination is also known as mixed-order, saturable, dose- or concentration-dependent, nonlinear, and Michaelis-Menten elimination.
Most drug elimination pathways will become saturated if the dose and therefore the concentration are high enough. When blood flow to an organ does not limit elimination , the relation between elimination rate and concentration (C) is expressed mathematically in equation (5):
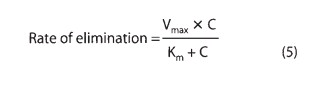
The maximum elimination capacity is Vmax, and Km is the drug concentration at which the rate of elimination is 50% of Vmax. At concentrations that are high relative to the Km, the elimination rate is almost independent of concentration—a state of “pseudo-zero order” elimination. If dosing rate exceeds elimination capac-ity, steady state cannot be achieved: The concentration will keep on rising as long as dosing continues. This pattern of capacity-limited elimination is important for three drugs in common use: ethanol, phenytoin, and aspirin. Clearance has no real meaning for drugs with capacity-limited elimination, and AUC should not be used to describe the elimination of such drugs.
B. Flow-Dependent Elimination
In contrast to capacity-limited drug elimination, some drugs are cleared very readily by the organ of elimination, so that at any clinically realistic concentration of the drug, most of the drug in the blood perfusing the organ is eliminated on the first pass of the drug through it. The elimination of these drugs will thus depend primarily on the rate of drug delivery to the organ of elimination. Such drugs (see Table 4–7) can be called “high-extraction” drugs since they are almost completely extracted from the blood by the organ. Blood flow to the organ is the main determinant of drug delivery, but plasma protein binding and blood cell partitioning may also be important for extensively bound drugs that are highly extracted.
Related Topics