Chapter: Basic & Clinical Pharmacology : Adrenocorticosteroids And Adrenocortical Antagonists
Synthesis Inhibitors & Glucocorticoid Antagonists
ANTAGONISTS OF ADRENOCORTICAL AGENTS
SYNTHESIS INHIBITORS &
GLUCOCORTICOID ANTAGONISTS
Inhibitors of steroid
synthesis act at several different steps and one glucocorticoid antagonist acts
at the receptor level.
Aminoglutethimide
Aminoglutethimide
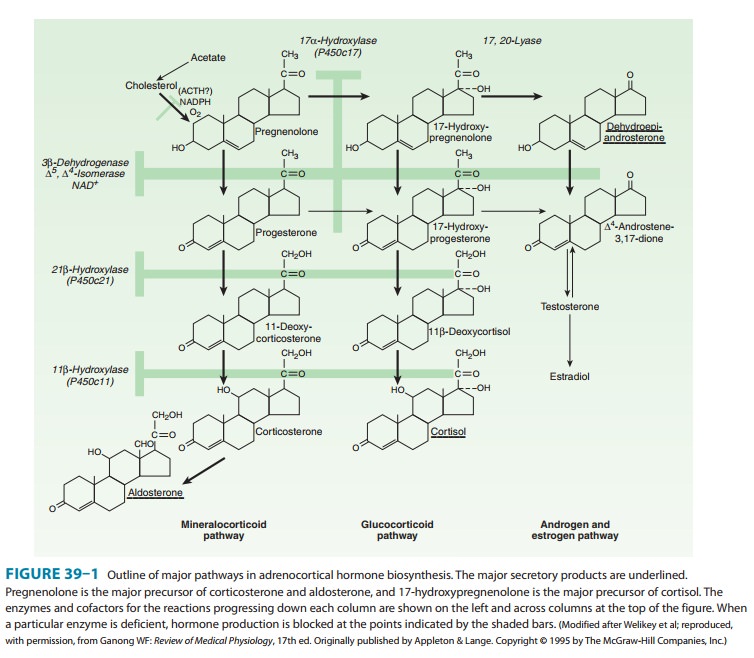
(Figure 39–5) blocks the conversion of cho-lesterol to pregnenolone (see Figure
39–1) and causes a reduction
It has been used in conjunction with
dexamethasone or hydrocortisone to reduce or eliminate estrogen production in
patients with carcinoma of the breast. In a dosage of 1 g/d it was well
tolerated; however, with higher dosages, lethargy and skin rash were common
effects. The use of aminoglutethimide in breast cancer patients has now been
supplanted by tamoxifen or by another class of drugs, the aro-matase
inhibitors. Aminoglutethimide can be used in conjunction with metyrapone or
ketoconazole to reduce steroid secretion in patients with Cushing’s syndrome
due to adrenocortical cancer who do not respond to mitotane.
Aminoglutethimide also
apparently increases the clearance of some steroids. It has been shown to
enhance the metabolism of dexamethasone, reducing its half-life from 4–5 hours
to 2 hours.
Ketoconazole
Ketoconazole, an antifungal imidazole derivative , is a potent and rather nonselective inhibitor of adrenal and gonadal steroid synthesis. This compound inhibits the cholesterol side-chain cleavage, P450c17, C17,20-lyase, 3β-hydroxysteroid dehydrogenase, and P450c11 enzymes required for steroid hor-mone synthesis. The sensitivity of the P450 enzymes to this com-pound in mammalian tissues is much lower than that needed to treat fungal infections, so that its inhibitory effects on steroid biosynthesis are seen only at high doses.
Ketoconazole has been
used for the treatment of patients with Cushing’s syndrome due to several
causes. Dosages of 200–1200 mg/d have produced a reduction in hormone levels
and clinical improvement in some patients. This drug has some hepatotoxicity
and should be started at 200 mg/d and slowly increased by 200 mg/d every 2–3
days up to a total daily dose of 1000 mg.
Metyrapone
Metyrapone (Figure
39–5) is a relatively selective inhibitor of steroid 11-hydroxylation,
interfering with cortisol and corticoster-one synthesis. In the presence of a
normal pituitary gland, there is a compensatory increase in pituitary ACTH
release and adrenal 11-deoxycortisol secretion. This response is a measure of
the capacity of the anterior pituitary to produce ACTH and has been adapted for
clinical use as a diagnostic test. Although the toxicity of metyrapone is much
lower than that of mitotane (see text that follows), the drug may produce
transient dizziness and gastroin-testinal disturbances. This agent has not been
widely used for the treatment of Cushing’s syndrome. However, in doses of 0.25
g twice daily to 1 g four times daily, metyrapone can reduce cortisol
production to normal levels in some patients with endogenous Cushing’s
syndrome. Thus, it may be useful in the management of severe manifestations of
cortisol excess while the cause of this condition is being determined or in
conjunction with radiation or surgical treatment. Metyrapone is the only
adrenal-inhibiting medication that can be administered to pregnant women with
Cushing’s syndrome. The major adverse effects observed are salt and water
retention and hirsutism resulting from diversion of the 11-deoxycortisol
precursor to DOC and androgen synthesis.
Metyrapone
is commonly used in tests of adrenal function. The blood levels of
11-deoxycortisol and the urinary excretion of 17-hydroxycorticoids are measured
before and after administra-tion of the compound. Normally, there is a twofold
or greater increase in the urinary 17-hydroxycorticoid excretion. A dose of
300–500 mg every 4 hours for six doses is often used, and urine collections are
made on the day before and the day after treatment. In patients with Cushing’s
syndrome, a normal response to metyrapone indicates that the cortisol excess is
not the result of a cortisol-secreting adrenal carcinoma or adenoma, since
secretion by such tumors produces suppression of ACTH and atrophy of normal
adrenal cortex.
Pituitary function may
also be tested by administering metyrapone, 2–3 g orally at midnight and by
measuring the level of ACTH or 11-deoxycortisol in blood drawn at 8 AM or by comparing
the excretion of 17-hydroxycorticosteroids in the urine during the 24-hour
periods preceding and following administra-tion of the drug. In patients with
suspected or known lesions of the pituitary, this procedure is a means of
estimating the ability of the gland to produce ACTH. Metyrapone has been
with-drawn from the market in the USA but is available on a compas-sionate
basis.
Trilostane
Trilostane is a 3β-17 hydroxysteroid
dehydrogenase inhibitor that interferes with the synthesis of adrenal and
gonadal hormones and is comparable to aminoglutethimide. Trilostane’s adverse
effects are predominantly gastrointestinal; adverse effects occur in about 50%
of patients with both trilostane and aminoglutethimide. There is no
cross-resistance or crossover of side effects between these compounds.
Trilostane is not available in the USA.
Abiraterone
Abiraterone is the
newest of the steroid synthesis inhibitors to enter clinical trials. It blocks
17α-hydroxylase
(P450c17) and 17,20-lyase (Figure 39–1), and predictably reduces synthesis of
cortisol and gonadal steroids in the adrenal and gonadal steroids in the
gonads. A compensatory increase occurs in ACTH and aldosterone synthesis, but
this can be prevented by concomitant administration of dexamethasone.
Abiraterone is an orally active steroid prodrug and has been studied in the
treatment of refrac-tory prostate cancer.
Mifepristone (RU-486)
The search for a
glucocorticoid receptor antagonist finally suc-ceeded in the early 1980s with
the development of the 11β-aminophenyl-substituted 19-norsteroid called
RU-486, later named mifepristone. Unlike the enzyme inhibitors previously
discussed, mifepristone is a pharmacologic antagonist at the ste-roid receptor.
This compound has strong antiprogestin activity and initially was proposed as a
contraceptive-contragestive agent. High doses of mifepristone exert
antiglucocorticoid activity by blocking the glucocorticoid receptor, since
mifepristone binds to it with high affinity, causing (1) some stabilization of
the hsp-glucocorticoid receptor complex and inhibition of the disso-ciation of
the RU-486–bound glucocorticoid receptor from the hsp chaperone proteins; and
(2) alteration of the interaction of the glucocorticoid receptor with
coregulators, favoring the formation of a transcriptionally inactive complex in
the cell nucleus. The result is inhibition of glucocorticoid receptor
activation.
The mean half-life of
mifepristone is 20 hours. This is longer than that of many natural and
synthetic glucocorticoid agonists (dexamethasone has a half-life of 4–5 hours).
Less than 1% of the daily dose is excreted in the urine, suggesting a minor
role of kid-neys in the clearance of the compound. The long plasma half-life of
mifepristone results from extensive and strong binding to plasma proteins. Less
than 5% of the compound is found in the free form when plasma is analyzed by
equilibrium dialysis. Mifepristone can bind to albumin and α1-acid glycoprotein,
but it has no affinity for corticosteroid-binding globulin.
In humans, mifepristone
causes generalized glucocorticoid resis-tance. Given orally to several patients
with Cushing’s syndrome due to ectopic ACTH production or adrenal carcinoma, it
was able to reverse the cushingoid phenotype, to eliminate carbohy-drate
intolerance, normalize blood pressure, to correct thyroid and gonadal hormone
suppression, and to ameliorate the psychological sequelae of hypercortisolism
in these patients. At present, this use of mifepristone can only be recommended
for inoperable patients with ectopic ACTH secretion or adrenal carcinoma who
have failed to respond to other therapeutic manipulations.
Mitotane
Mitotane
(Figure 39–5), a drug related to the DDT class of insec-ticides, has a
nonselective cytotoxic action on the adrenal cortex in dogs and to a lesser
extent in humans. This drug is administered orally in divided doses up to 12 g
daily. About one third of patients with adrenal carcinoma show a reduction in
tumor mass. In 80% of patients, the toxic effects are sufficiently severe to
require dose reduction. These include diarrhea, nausea, vomiting, depression,
somnolence, and skin rashes. The drug has been with-drawn from the market in
the USA but is available on a compas-sionate basis
Related Topics