Chapter: Basic & Clinical Pharmacology : Adrenocorticosteroids And Adrenocortical Antagonists
Clinical Pharmacology - Synthetic Corticosteroids
CLINICAL PHARMACOLOGY
A. Diagnosis and Treatment of Disturbed Adrenal Function
1. Adrenocortical insufficiency
Chronic (Addison’s disease)—Chronic adrenocortical insuffi-ciency is characterized by weakness, fatigue, weight loss, hypoten-sion, hyperpigmentation, and inability to maintain the blood glucose level during fasting. In such individuals, minor noxious, traumatic, or infectious stimuli may produce acute adrenal insuffi-ciency with circulatory shock and even death.
In primary adrenal
insufficiency, about 20–30 mg of hydrocor-tisone must be given daily, with
increased amounts during periods of stress. Although hydrocortisone has some
mineralocorticoid activity, this must be supplemented by an appropriate amount
of a salt-retaining hormone such as fludrocortisone. Synthetic gluco-corticoids
that are long-acting and devoid of salt-retaining activity should not be
administered to these patients.
Acute—When acute
adrenocortical insufficiency is suspected,treatment must be instituted
immediately. Therapy consists of large amounts of parenteral hydrocortisone in
addition to correc-tion of fluid and electrolyte abnormalities and treatment of
pre-cipitating factors.
Hydrocortisone
sodium succinate or phosphate in doses of 100 mg intravenously is given every 8
hours until the patient is stable. The dose is then gradually reduced,
achieving maintenance dosage within 5 days.
The
administration of salt-retaining hormone is resumed when the total
hydrocortisone dosage has been reduced to 50 mg/d.
2. Adrenocortical hypo- and hyperfunction
·
Congenital adrenal
hyperplasia—This
group of disorders ischaracterized by specific defects in the synthesis of
cortisol. In pregnancies at high risk for congenital adrenal hyperplasia,
fetuses can be protected from genital abnormalities by administrationof
dexamethasone to the mother. The most common defect is a decrease in or lack of
P450c21 (21β-hydroxylase)
activity.∗
As
can be seen in Figure 39–1, this would lead to a reduction in cortisol
synthesis and thus produce a compensatory increase in ACTH release. The gland
becomes hyperplastic and produces ab-normally large amounts of precursors such
as 17-hydroxyproges-terone that can be diverted to the androgen pathway,
leading to virilization. Metabolism of this compound in the liver leads to
pregnanetriol, which is characteristically excreted into the urine in large
amounts in this disorder and can be used to make the diag-nosis and to monitor
efficacy of glucocorticoid substitution. However, the most reliable method of
detecting this disorder is the increased response of plasma
17-hydroxyprogesterone to ACTH stimulation.
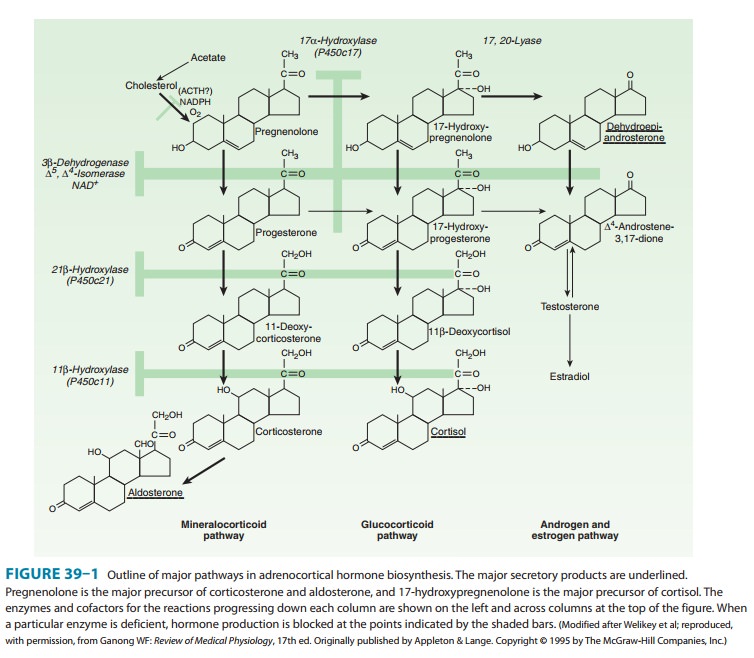
If the defect is in
11-hydroxylation, large amounts of deoxycor-ticosterone are produced, and
because this steroid has mineralocor-ticoid activity, hypertension with or
without hypokalemic alkalosis ensues. When 17-hydroxylation is defective in the
adrenals and gonads, hypogonadism is also present. However, increased amounts
of 11-deoxycorticosterone are formed, and the signs and symptoms associated
with mineralocorticoid excess—such as hypertension and hypokalemia—are also
observed.
When
first seen, the infant with congenital adrenal hyperplasia may be in acute
adrenal crisis and should be treated as described above, using appropriate
electrolyte solutions and an intravenous preparation of hydrocortisone in
stress doses.
Once
the patient is stabilized, oral hydrocortisone, 12–18 mg/m2/d
in two unequally divided doses (two thirds in the morning, one third in late
afternoon) is begun. The dosage is adjusted to allow normal growth and bone
maturation and to prevent androgen excess. Alternate-day therapy with
prednisone has also been used to achieve greater ACTH suppression without
increasing growth inhi-bition. Fludrocortisone, 0.05–0.2 mg/d, should also be
adminis-tered by mouth, with added salt to maintain normal blood pressure,
plasma renin activity, and electrolytes.
Cushing’s syndrome—Cushing’s
syndrome is usually the resultof bilateral adrenal hyperplasia secondary to an
ACTH-secreting pituitary adenoma (Cushing’s disease) but occasionally is due to
tumors or nodular hyperplasia of the adrenal gland or ectopic pro-duction of
ACTH by other tumors. The manifestations are those associated with the chronic
presence of excessive glucocorticoids. When glucocorticoid hypersecretion is
marked and prolonged, a rounded, plethoric face and trunk obesity are striking
in appear-ance. The manifestations of protein loss are often found and include
muscle wasting; thinning, purple striae, and easy bruising of the skin; poor
wound healing; and osteoporosis. Other serious disturbances include mental
disorders, hypertension, and diabetes. This disorder is treated by surgical
removal of the tumor produc-ing ACTH or cortisol, irradiation of the pituitary
tumor, or resec-tion of one or both adrenals. These patients must receive large
doses of cortisol during and after the surgical procedure. Doses of up to 300
mg of soluble hydrocortisone may be given as a continu-ous intravenous infusion
on the day of surgery. The dose must be reduced slowly to normal replacement
levels, since rapid reduction in dose may produce withdrawal symptoms,
including fever and joint pain. If adrenalectomy has been performed, long-term
main-tenance is similar to that outlined above for adrenal insufficiency.
c. Primary
generalized glucocorticoid resistance (Chrousos) syndrome—This rare sporadic or
familial genetic condition isusually due to inactivating mutations of the
glucocorticoid recep-tor gene. In its attempt to compensate for the defect, the
hypo-thalamic-pituitary-adrenal (HPA) axis is hyperfunctioning with the
increased production of ACTH leading to high circulating levels of cortisol and
cortisol precursors such as corticosterone and 11-deoxycorticosterone with
mineralocorticoid activity, as well as of adrenal androgens. These may result
in hypertension with or without hypokalemic alkalosis and hyperandrogenism
expressed as virilization and precocious puberty in children and acne,
hir-sutism, male pattern baldness, and menstrual irregularities (mostly
oligo-amenorrhea and hypofertility) in women. The therapy of this syndrome is
high doses of synthetic glucocorticoids such as dexamethasone with no inherent
mineralocorticoid activity. These doses are titrated to normalize the
production of cortisol, cortisol precursors, and adrenal androgens.
d. Aldosteronism—Primary
aldosteronism usually results fromthe excessive production of aldosterone by an
adrenal adenoma. However, it may also result from abnormal secretion by
hyperplasticglands or from a malignant tumor. The clinical findings of
hyper-tension, weakness, and tetany are related to the continued renal loss of
potassium, which leads to hypokalemia, alkalosis, and elevation of serum sodium
concentrations. This syndrome can also be produced in disorders of adrenal
steroid biosynthesis by excessive secretion of deoxycorticosterone,
corticosterone, or 18-hydroxycorticosterone— all compounds with inherent
mineralocorticoid activity.
In contrast to
patients with secondary aldosteronism (see text that follows), these patients
have low (suppressed) levels of plasma renin activity and angiotensin II. When
treated with fludrocorti-sone (0.2 mg twice daily orally for 3 days) or
deoxycorticosterone acetate (20 mg/d intramuscularly for 3 days—but not
available in the USA), patients fail to retain sodium and the secretion of
aldos-terone is not significantly reduced. When the disorder is mild, it may
escape detection if serum potassium levels are used for screen-ing. However, it
may be detected by an increased ratio of plasma aldosterone to renin. Patients
generally improve when treated with spironolactone, an aldosterone
receptor-blocking agent, and the response to this agent is of diagnostic and
therapeutic value.
3. Use of glucocorticoids for
diagnostic purposes—It issometimes necessary to suppress the production of ACTH to
iden-tify the source of a particular hormone or to establish whether its
production is influenced by the secretion of ACTH. In these cir-cumstances, it
is advantageous to use a very potent substance such as dexamethasone because
the use of small quantities reduces the possibility of confusion in the
interpretation of hormone assays in blood or urine. For example, if complete
suppression is achieved by the use of 50 mg of cortisol, the urinary
17-hydroxycorticosteroids will be 15–18 mg/24 h, since one third of the dose
given will be recovered in urine as 17-hydroxycorticosteroid. If an equivalent
dose of 1.5 mg of dexamethasone is used, the urinary excretion will be only 0.5
mg/24 h and blood levels will be low.
The dexamethasone suppression test is used
for the diagnosis of Cushing’s syndrome and has also been used in the differential
diagnosis of depressive psychiatric states. As a screening test, 1 mg
dexamethasone is given orally at 11 PM, and a plasma sample is obtained the
following morning. In normal individuals, the morn-ing cortisol concentration
is usually less than 3 mcg/dL, whereas in Cushing’s syndrome the level is
usually greater than 5 mcg/dL. The results are not reliable in the patient with
depression, anxiety, con-current illness, and other stressful conditions or in
the patient who is receiving a medication that enhances the catabolism of
dexame-thasone in the liver. To distinguish between hypercortisolism due to
anxiety, depression, and alcoholism (pseudo-Cushing syndrome) and bona fide
Cushing’s syndrome, a combined test is carried out, consisting of dexamethasone
(0.5 mg orally every 6 hours for 2 days) followed by a standard
corticotropin-releasing hormone (CRH) test (1 mg/kg given as a bolus
intravenous infusion 2 hours after the last dose of dexamethasone).
In patients in whom
the diagnosis of Cushing’s syndrome has been established clinically and
confirmed by a finding of elevated free cortisol in the urine, suppression with
large doses of dexa-methasone will help to distinguish patients with Cushing’s
disease from those with steroid-producing tumors of the adrenal cortex or with
the ectopic ACTH syndrome. Dexamethasone is given in adosage of 0.5 mg orally
every 6 hours for 2 days, followed by 2 mg orally every 6 hours for 2 days, and
the urine is then assayed for cortisol or its metabolites (Liddle’s test); or
dexamethasone is given as a single dose of 8 mg at 11 PM, and the plasma
cortisol is mea-sured at 8 AM the following day. In patients with Cushing’s
dis-ease, the suppressant effect of dexamethasone usually produces a 50%
reduction in hormone levels. In patients in whom suppres-sion does not occur,
the ACTH level will be low in the presence of a cortisol-producing adrenal
tumor and elevated in patients with an ectopic ACTH-producing tumor.
B. Corticosteroids and Stimulation of Lung Maturation in the Fetus
Lung
maturation in the fetus is regulated by the fetal secretion of cortisol.
Treatment of the mother with large doses of glucocorti-coid reduces the
incidence of respiratory distress syndrome in infants delivered prematurely.
When delivery is anticipated before 34 weeks of gestation, intramuscular
betamethasone, 12 mg, followed by an additional dose of 12 mg 18–24 hours later, is
commonly used. Betamethasone is chosen because maternal
pro-tein binding and placental metabolism of this corticosteroid is less than
that of cortisol, allowing increased transfer across the placenta to the fetus.
C. Corticosteroids and Nonadrenal Disorders
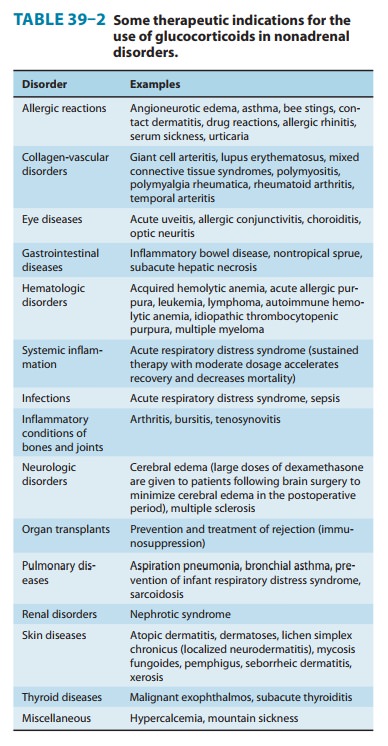
The
synthetic analogs of cortisol are useful in the treatment of a diverse group of
diseases unrelated to any known disturbance of adrenal function (Table 39–2).
The usefulness of corticosteroids in these disorders is a function of their
ability to suppress inflamma-tory and immune responses and to alter leukocyte
function, as previously described. These agents are useful in disorders in
which host response is the cause of the major manifestations of the disease. In
instances in which the inflammatory or immune response is important in
controlling the pathologic process, therapy with corticosteroids may be
dangerous but justified to prevent irreparable damage from an inflammatory
response—if used in conjunction with specific therapy for the disease process.
Since corticosteroids
are not usually curative, the pathologic process may progress while clinical
manifestations are suppressed. Therefore, chronic therapy with these drugs
should be undertaken with great care and only when the seriousness of the
disorder warrants their use and when less hazardous measures have been
exhausted.
In general, attempts should be made to bring the disease process under control using medium- to intermediate-acting glucocorti-coids such as prednisone and prednisolone (Table 39–1), as well as all ancillary measures possible to keep the dose low. Where possible, alternate-day therapy should be used (see the following text). Therapy should not be decreased or stopped abruptly. When pro-longed therapy is anticipated, it is helpful to obtain chest x-rays and a tuberculin test, since glucocorticoid therapy can reactivate dormant tuberculosis. The presence of diabetes, peptic ulcer, osteo-porosis, and psychological disturbances should be taken into con-sideration, and cardiovascular function should be assessed.
Treatment for transplant
rejection is a very important applica-tion of glucocorticoids. The efficacy of
these agents is based on their ability to reduce antigen expression from the
grafted tissue, delay revascularization, and interfere with the sensitization
of cytotoxic T lymphocytes and the generation of primary antibody-forming
cells.
Related Topics