Chapter: Essential Microbiology: Microorganisms in Genetic Engineering
Plasmid cloning vectors
Plasmid
cloning vectors
Two main types of vector system are used in cloning,
those that use plasmids and those
that use bacterio-phages. Naturally
occur-ring examples of these are manipulated so that they pos-sess the above
properties. A popular vector in the early days of gene cloning was the plasmid
pBR322; Figure 12.4 shows how it contains the features described above.

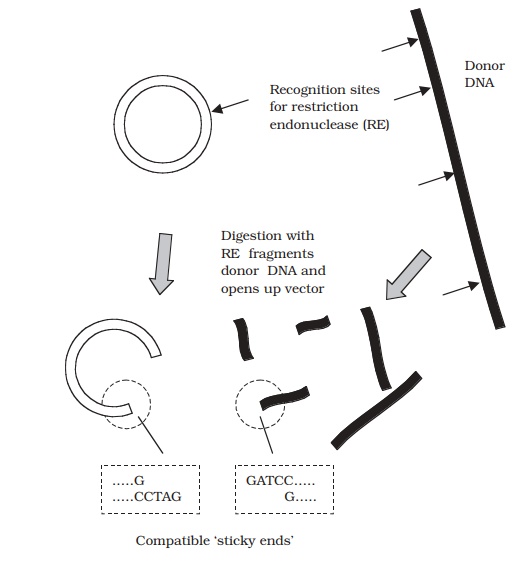
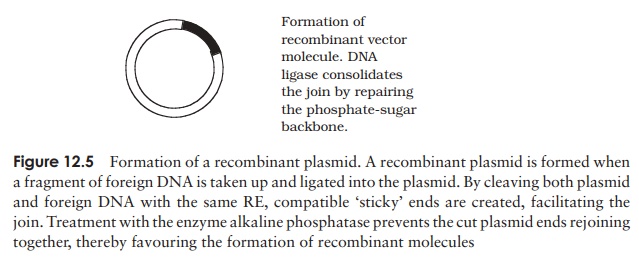
Let us consider now what happens, at a molecular
level, when donor DNA is ligated into a plasmid vector (Figure 12.5). Sticky
ends of the donor fragment form hy-drogen bonds with the exposed compatible
ends of the opened up plasmid by complementary base pairing, and DNA ligase
consolidates the join. Many plasmids con-tain engineered sequences called multiple cloning sites (MCS); these
provide additional flexibility with respect to the restriction fragments that
may be accommodated.
The ligation of insert DNA into the cloning site of
the plasmid is not the only possible outcome of the pro-cedure described above,
however. Unless experimental conditions are carefully controlled (there are
ways of doing this), a more likely outcome is that the two com-patible ends of
the plasmid will simply ‘find’ each other
again, and rejoin. Since a certain amount of this is
inevitable, how are we able to tell the difference between those bacteria that
contain a recombinant plasmid (one containing a piece of donor DNA) and those
that have taken up a recircularised ‘native’ plasmid? Since both types will
contain the gene for ampicillin resistance, we cannot distinguish them by this
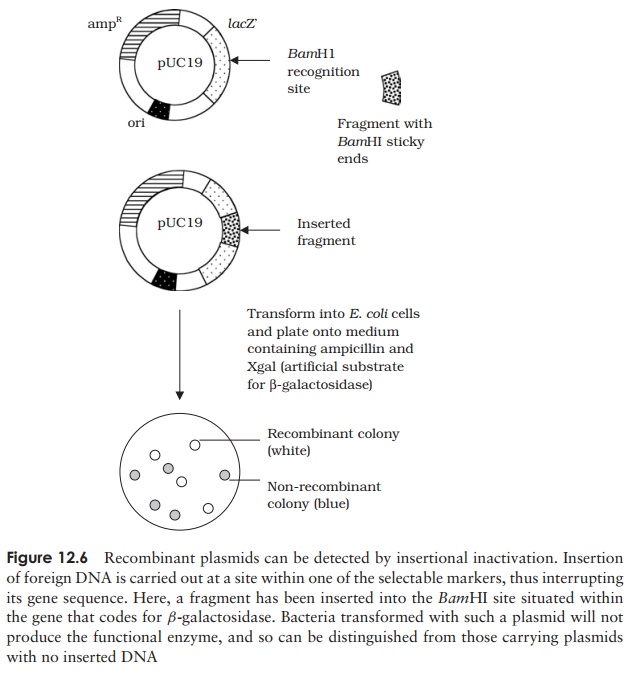
means. A strategy commonly used to get around the problem is insertional inactivation (Figure 12.6).
This clever ploy exploits the fact that we can manipulate DNA, and, for
example, insert RE recognition sequences at desired points. If a recognition
site occurs in the middle of a gene sequence, and a piece of foreign DNA is
inserted at this position, the gene will be interrupted, and unable to produce
a functional gene product. In the example shown, the gene is lacZ , necessary for the successful expression of the enzyme β-galactosidase. This will only be expressed in those
bacteria that contain plasmids in which the gene has remained uninterrupted,
i.e. those that have not taken up an
insert. Expression of the β-galactosidase
can be detected by growing the bacteria on an artificial substrate, which is
converted to a coloured (usually blue) product when acted on by the enzyme.
Those cells that contain recombinant plasmids are easily identified because
disruption of the lacZ gene means
that no β-galactosidase
is produced, resulting in non-pigmented (white) colonies.
One problem remains. Remember that our inserted DNA
was derived from the di-gestion of total (genomic) DNA from the donor organism;
this means that our DNA library will contain recombinant plasmids with a whole
range of fragments from that digestion, and not just the specific one that
interests us. How are we able to distinguish this fragment from the others?
A technique called nucleic acid hybridisation is used to solve the problem. This once
again depends on complementary base pairing, and involves the creation of a probe, a short length of single-stranded
DNA that is complementary to part of the desired ‘target’ sequence, and
therefore able to seek it out. If searching for the right clone can be likened
to looking for a needle in a haystack, then the probe is a powerful ‘magnet’
that makes the task much easier. The probe carries a tag or label, so that its
location can be identified (Figure 12.7).

Once we have identified the clone of bacteria
containing plasmids with the insert that interests us, we can grow a pure
culture of it and then isolate plasmid DNA. Using the same RE as before, the
inserted donor DNA can be removed and purified. We now have enough of this
specific DNA sequence (a tiny proportion of the donor organism’s total genome)
to analyse and manipulate.
Although plasmids are easily isolated and
manipulated, their use as cloning vectors is limited by the fact that they tend
to become unstable if we attempt to insert much more than about 5kb of foreign
DNA. For inserts larger than this, we must turn to other vector systems.
Related Topics