Chapter: Basic & Clinical Pharmacology : The Alcohols
Pharmacodynamics of Acute Ethanol Consumption
Pharmacodynamics of Acute Ethanol
Consumption
A. Central Nervous System
The
CNS is markedly affected by acute alcohol consumption. Alcohol causes sedation,
relief of anxiety and, at higher concentra-tions, slurred speech, ataxia,
impaired judgment, and disinhibited behavior, a condition usually called
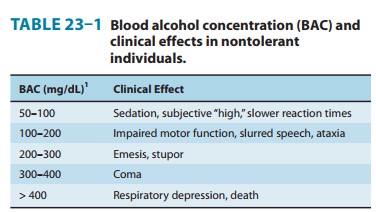
intoxication or drunkenness (Table 23–1). These CNS effects are most marked as
the blood level is rising, because acute tolerance to the effects of alcohol
occurs after a few hours of drinking. For chronic drinkers who are tolerant to
the effects of alcohol, higher concentrations are needed to elicit these CNS
effects. For example, an individual with chronic alcoholism may appear sober or
only slightly intoxicated with a blood alcohol concentration of 300–400 mg/dL,
whereas this level is associated with marked intoxication or even coma in a
nontoler-ant individual. The propensity of moderate doses of alcohol to inhibit
the attention and information-processing skills as well as the motor skills
required for operation of motor vehicles has pro-found effects. Approximately
30–40% of all traffic accidents resulting in a fatality in the United States
involve at least one person with blood alcohol near or above the legal level of
intoxication, and drunken driving is a leading cause of death in young adults.
Like
other sedative-hypnotic drugs, alcohol is a CNS depres-sant. At high blood
concentrations, it induces coma, respiratory depression, and death.
Ethanol
affects a large number of membrane proteins that participate in signaling
pathways, including neurotransmitter receptors for amines, amino acids,
opioids, and neuropeptides; enzymes such as Na+/K+-ATPase,
adenylyl cyclase, phosphoinositide-specific phospholipase C; a nucleoside
transporter; and ion channels. Much attention has focused on alcohol’s effects
on neurotransmis-sion by glutamate and γ-aminobutyric acid (GABA), the main excitatory
and inhibitory neurotransmitters in the CNS. Acute ethanol exposure enhances
the action of GABA at GABAA recep-tors, which is consistent with the
ability of GABA-mimetics to intensify many of the acute effects of alcohol and
of GABAA antagonists to attenuate some of the actions of ethanol.
Ethanol inhibits the ability of glutamate to open the cation channel
associ-ated with the N-methyl-D-aspartate
(NMDA) subtype of gluta-mate receptors. The NMDA receptor is implicated in many
aspects of cognitive function, including learning and memory.
“Blackouts”—periods of memory loss that occur with high levels of alcohol—may
result from inhibition of NMDA receptor activa-tion. Experiments that use
modern genetic approaches eventually will yield a more precise definition of
ethanol’s direct and indirect targets. In recent years, experiments with mutant
strains of mice, worms, and flies have reinforced the importance of previously
identified targets and helped identify new candidates, including a
calcium-regulated and voltage-gated potassium channel that may be one of
ethanol’s direct targets (see Box: What Can Drunken Worms, Flies, and Mice Tell
Us about Alcohol?).
B. Heart
Significant
depression of myocardial contractility has been observed in individuals who
acutely consume moderate amounts of alcohol, ie, at a blood concentration above
100 mg/dL.
C. Smooth Muscle
Ethanol
is a vasodilator, probably as a result of both CNS effects (depression of the
vasomotor center) and direct smooth muscle relaxation caused by its metabolite,
acetaldehyde. In cases of severe overdose, hypothermia—caused by
vasodilation—may be marked in cold environments. Ethanol also relaxes the
uterus and—before the introduction of more effective and safer uterine
relaxants (eg, calcium channel antagonists)—was used intravenously for the
suppression of premature labor.
Related Topics