Chapter: Basic & Clinical Pharmacology : Drugs Used in Heart Failure
Pathophysiology of Heart Failure
Pathophysiology of Heart Failure
Heart failure is a syndrome with many causes that may involve one or both ventricles. Cardiac output is usually below the normal range (“low-output” failure). Systolic dysfunction, with reducedcardiac output and significantly reduced ejection fraction (< 45%; normal > 60%), is typical of acute failure, especially that resulting from myocardial infarction. Diastolic dysfunction often occurs as a result of hypertrophy and stiffening of the myocardium, and although cardiac output is reduced, ejection fraction may be nor-mal. Heart failure due to diastolic dysfunction does not usually respond optimally to positive inotropic drugs.
“High-output”
failure is a rare form of heart failure. In this condition, the demands of the
body are so great that even increased cardiac output is insufficient.
High-output failure can result from hyperthyroidism, beriberi, anemia, and
arteriovenous shunts. This form of failure responds poorly to the drugs
discussed in this chap-ter and should be treated by correcting the underlying
cause.
The
primary signs and symptoms of all types of heart failure include tachycardia,
decreased exercise tolerance, shortness of breath, and cardiomegaly. Peripheral
and pulmonary edema (the congestion of congestive heart failure) are often but
not always present. Decreased exercise tolerance with rapid muscular fatigue is
the major direct consequence of diminished cardiac output. The other
manifestations result from the attempts by the body to com-pensate for the
intrinsic cardiac defect.
Neurohumoral (extrinsic) compensation involves two major
mechanisms (previously presented in Figure 6–7)—the sympa-thetic nervous system
and the renin-angiotensin-aldosterone hor-monal response—plus several others.
Some of the detrimental as well as beneficial features of these compensatory
responses are illustrated in Figure 13–2. The baroreceptor reflex appears to be
reset, with a lower sensitivity to arterial pressure, in patients with
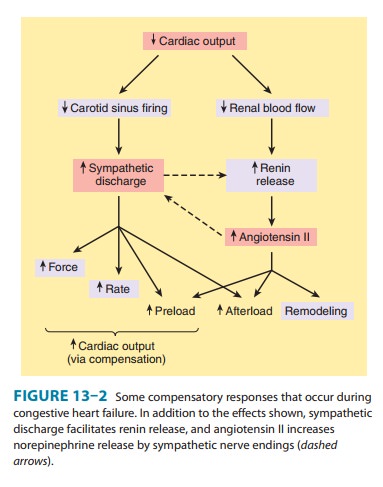
As a result, baroreceptor sensory input to the vaso-motor center is
reduced even at normal pressures; sympathetic outflow is increased, and
parasympathetic outflow is decreased. Increased sympathetic outflow causes
tachycardia, increased car-diac contractility, and increased vascular tone.
Vascular tone is further increased by angiotensin II and endothelin, a potent
vaso-constrictor released by vascular endothelial cells. Vasoconstriction
increases afterload, which further reduces ejection fraction and cardiac
output. The result is a vicious cycle that is characteristic of heart failure
(Figure 13–3). Neurohumoral antagonists and vaso-dilators reduce heart failure
mortality by interrupting the cycle and slowing the downward spiral.
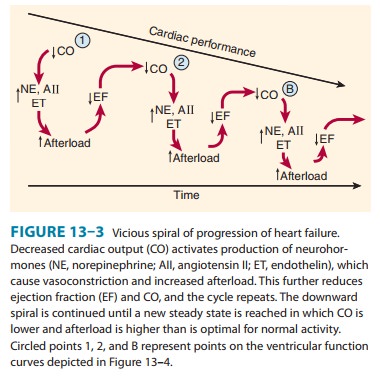
After
a relatively short exposure to increased sympathetic drive, complex
down-regulatory changes in the cardiac β1-adrenoceptor– G protein-effector system take
place that result in diminished stimulatory effects. Beta2 receptors
are not down-regulated and may
develop increased coupling to the IP3-DAG cascade. It has also been
suggested that cardiac β3 receptors (which do not appear to be
down-regulated in failure) may mediate negative
inotropic effects. Excessive β activation can lead to leakage of calcium
from the SR via RyR channels and contributes to stiffening of the ven-tricles
and arrhythmias. Prolonged β activation also increases cas-pases, the
enzymes responsible for apoptosis. Increased angiotensinproduction leads to
increased aldosterone secretion (with sodium and water retention), to increased
afterload, and to remodeling of both heart and vessels (discussed below). Other
hormones are released, including natriuretic peptide, endothelin, and
vasopressin . Within the heart, failure-induced changes have been documented in
calcium handling in the SR by SERCA and phospholamban; in transcription factors
that lead to hypertrophy and fibrosis; in mitochondrial function, which is
critical for energy production in the overworked heart; and in ion channels,
especially potassium channels, which facili-tate arrhythmogenesis, a primary
cause of death in heart failure. Phosphorylation of RyR channels in the
sarcoplasmic reticulum enhances and dephosphorylation reduces Ca2+ release; studies in
animal models indicate that the enzyme primarily responsible for RyR
dephosphorylation, protein phosphatase 1 (PP1), is up-regulated in heart
failure. These cellular changes provide many potential targets for future
drugs.
The
most important intrinsic compensatory mechanism is myocardial hypertrophy. This increase in muscle mass helpsmaintain
cardiac performance. However, after an initial beneficial effect, hypertrophy
can lead to ischemic changes, impairment of diastolic filling, and alterations
in ventricular geometry. Remodeling is
the term applied to dilation (other than that due topassive stretch) and other
slow structural changes that occur in the stressed myocardium. It may include
proliferation of connective tissue cells as well as abnormal myocardial cells
with some bio-chemical characteristics of fetal myocytes. Ultimately, myocytes
in the failing heart die at an accelerated rate through apoptosis, leav-ing the
remaining myocytes subject to even greater stress.
Related Topics