Chapter: Basic & Clinical Pharmacology : Immunopharmacology
Normal Immune Responses
ELEMENTS OF THE IMMUNE SYSTEM
NORMAL IMMUNE RESPONSES
The
immune system has evolved to protect the host from invading pathogens and to
eliminate disease. At its functioning best, the immune system is exquisitely
responsive to invading pathogens while retaining the capacity to recognize self
tissues and antigens to which it is tolerant. Protection from infection and
disease is provided by the collaborative efforts of the innate and adaptive
immune systems.
The Innate Immune System
The innate immune
system is the first line of defense against invading pathogens (eg, bacteria,
viruses, fungi, parasites) and consists of mechanical, biochemical, and
cellular components. Mechanical components include skin/epidermis and mucus; bio-chemical
components include antimicrobial peptides and proteins (eg, defensins),
complement, enzymes (eg, lysozyme, acid hydro-lases), interferons, acidic pH,
and free radicals (eg, hydrogen per-oxide, superoxide anions); cellular
components include neutrophils, monocytes, macrophages, natural killer (NK),
and natural killer-T (NKT) cells. Unlike adaptive immunity, the innate immune
response exists prior to infection, is not enhanced by repeated infection, and
is generally not antigen-specific. An intact skin or mucosa is the first
barrier to infection. When this barrier is breached, an immediate innate immune
response, referred to as “inflammation” is provoked that ultimately leads to
destruction of the pathogen. The process of pathogen destruction can be
accom-plished, for example, by biochemical components such as lysozyme (which
breaks down the protective peptidoglycan cell wall) and the split products
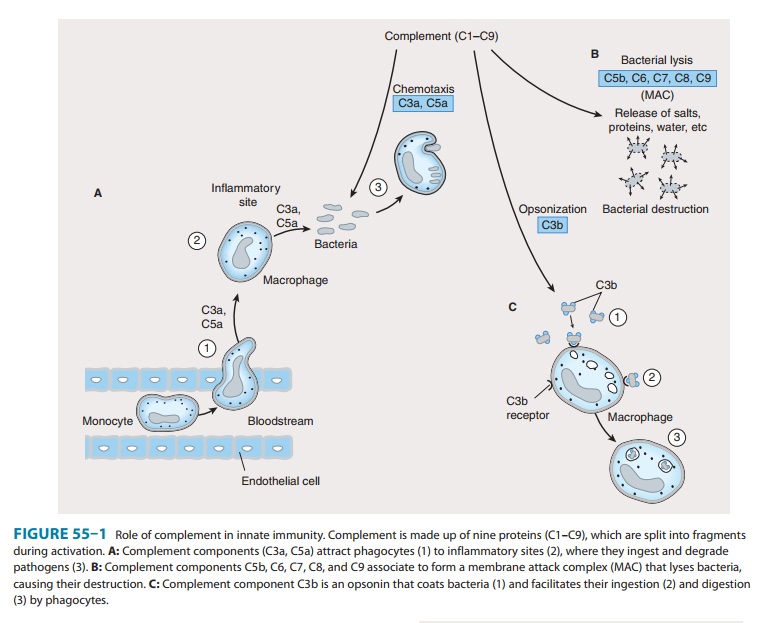
arising from complement activation. Complement components (Figure 55–1) enhance
macrophage and neutrophil phagocytosis by acting as opsonins (C3b) and
chemoattractants (C3a, C5a), which recruit immune cells from the bloodstream to
the site of infection. The activation of comple-ment eventually leads to
pathogen lysis via the generation of a membrane attack complex that creates
holes in the membrane and results in leakage of cellular components.
During the inflammatory response triggered by infection, neu-trophils and monocytes enter the tissue sites from peripheral circula-tion. This cellular influx is mediated by the action of chemoattractant cytokines (chemokines) (eg, interleukin-8 [IL-8;CXCL8], macrophage chemotactic protein-1 [MCP-1; CCL2], and macrophage inflammatory protein-1α [MIP-1α; CCL3]) released from activated endothelial cells and immune cells (mostly tissue macrophages) at the inflammatory site.
Egress of the immune cells from blood vessels into the inflammatory site is
mediated by adhesive interactions between cell surface receptors (eg, L-selectin, integrins)
on the immune cells and ligands (eg, sialyl-Lewis x, intercellular adhesion
molecule-1 [ICAM-1]) on the activated endothelial cell surface. The tissue
macrophages as well as dendritic cells express pattern recognition receptors
(PRRs) that include Toll-like receptors (TLRs), nucleotide-binding
oli-gomerization domain (NOD)-like receptors (NLRs), scavenger receptors,
mannose receptors, and lipopolysaccharide (LPS)-binding protein, which
recognize key evolutionarily conserved pathogen components referred to as
pathogen-associated molecu-lar patterns (PAMPs). Examples of PAMPs include
microbe-derived unmethylated CpG DNA, flagellin, double-stranded RNA,
peptidoglycan, and LPS. The PRRs recognize PAMPs in various components of
pathogens and stimulate the release of proinflammatory cytokines, chemokines,
and interferons. If the innate immune response is successfully executed, the
invading pathogen is ingested, degraded, and eliminated, and disease is either
prevented or is of short duration.
In addition to
monocytes and neutrophils, natural
killer (NK),natural killer-T (NKT), and
gamma-delta T (fc T) cells recruitedto the inflammatory site contribute to the innate
response by secret-ing interferon-gamma (IFN-γ) and IL-17, which activate resident tissue
macrophages and dendritic cells and recruit neutrophils respectively to
successfully eliminate invading pathogens. NK cells are so called because they
are able to recognize and destroy virus-infected normal cells as well as tumor
cells without prior stimula-tion. This activity is regulated by “killer cell
immunoglobulin-like receptors” (KIRs) on the NK cell surface that are specific
for major histocompatibility complex (MHC) class I molecules. When NK cells
bind self MHC class I proteins (expressed on all nucleated cells), these
receptors deliver inhibitory signals, preventing them from killing normal host
cells. Tumor cells or virus-infected cells that have down-regulated MHC class I
expression do not engage these KIRs, resulting in activation of NK cells and
subsequent destruction of the target cell. NK cells kill target cells by
releasing cytotoxic granules such as perforins and granzymes that induce
programmed cell death.
NKT
cells express T-cell receptors as well as receptors com-monly found on NK
cells. NKT cells recognize microbial lipid antigens presented by a unique class
of MHC-like molecules known as CD1 and have been implicated in host defense
against microbial agents, autoimmune diseases, and tumors.
The Adaptive Immune System
The adaptive immune
system is mobilized by cues from the innate response when the innate processes
are incapable of coping with an infection. The adaptive immune system has a
number of char-acteristics that contribute to its success in eliminating pathogens.
These include the ability to (1) respond to a variety of antigens, each in a
specific manner; (2) discriminate between foreign (“non-self ”) antigens
(pathogens) and self antigens of the host; and (3) respond to a previously
encountered antigen in a learned way by initiating a vigorous memory response.
This adaptive response culminates in the production of antibodies, which are the effec-tors of humoral immunity; and the activation of T lymphocytes, which are the effectors of cell-mediated immunity.
The induction of
specific adaptive immunity requires the par-ticipation of professional antigen-presenting cells (APCs), which
include dendritic cells (DCs), macrophages, and B lymphocytes. These cells play
pivotal roles in the induction of an adaptive immune response because of their
capacity to phagocytize particu-late antigens (eg, pathogens) or endocytose
protein antigens, and enzymatically digest them to generate peptides, which are
then loaded onto class I or class II MHC proteins and “presented” to the cell
surface T-cell receptor (TCR) (Figure 55–2). CD8 T cells recognize class I-MHC
peptide complexes while CD4 T cells rec-ognize class II-MHC peptide complexes.
At least two signals are necessary for the activation of T cells. The first
signal is delivered following engagement of the TCR with peptide-bound MHC
molecules. In the absence of a second signal, the T cells become unresponsive
(anergic) or undergo apoptosis. The second signal involves ligation of
costimulatory molecules (CD40, CD80 [also known as B7-1], and CD86 [also known
as B7-2]) on the APC to
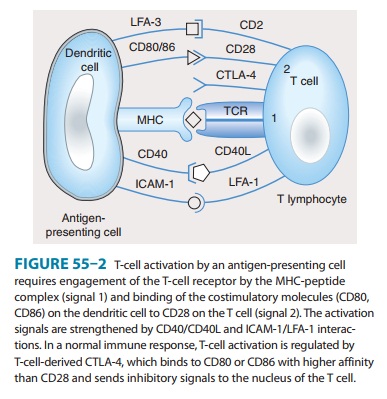
their respective
ligands (CD40L for CD40, CD28 for CD80 or CD86). Activation of T cells is
regulated via a negative feedback loop involving another molecule known as
T-lymphocyte-associated antigen 4 (CTLA-4). Following engagement of CD28 with
CD80 or CD86, CTLA-4 in the cytoplasm is mobilized to the cell surface where,
because of its higher affinity of binding to CD80 and CD86, it outcompetes or displaces
CD28 resulting in suppression of T-cell activation and proliferation. This
property of CTLA-4 has been exploited as a strategy for sustaining a desirable
immune response such as that directed against cancer. A recombi-nant humanized
antibody that binds CTLA-4 (ipilimumab) pre-vents its association with
CD80/CD86. In so doing, the activated state of T cells is sustained. Recently
completed vaccine trials in metastatic melanoma patients receiving anti-CTLA-4
antibody reported objective and durable clinical responses in a few patients.
Unfortunately, these beneficial responses were associated with the development
of autoimmune toxicity in some patients, raising concern about this approach.
T lymphocytes develop and learn to recognize self and non-self antigens in the thymus; those T cells that bind with high affinity to self antigens in the thymus undergo apoptosis (negative selection), while those that are capable of recognizing foreign antigens in the presence of self MHC molecules are retained and expanded (posi-tive selection) for export to the periphery (lymph nodes, spleen, mucosa-associated lymphoid tissue, peripheral blood), where they become activated after encountering MHC-presented peptides (Figures 55–2 and 55–3).
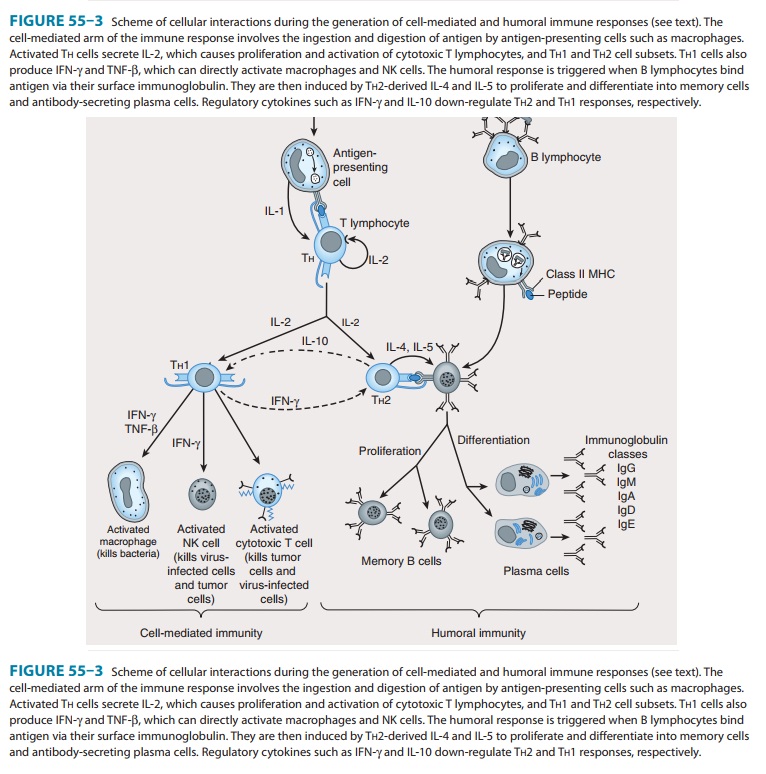
Studies using murine
T-cell clones have demonstrated the pres-ence of two subsets of T helper
lymphocytes (TH1 and TH2) based on the cytokines they secrete after activation. This
demarcation is not so clear-cut in humans. The TH1 subset characteristicallyproduces IFN-γ, IL-2, and IL-12 and induces cell-mediated
immunity by activation of macrophages, cytotoxic T cells (CTLs), and NK cells.
The TH2 subset produces
IL-4, IL-5, IL-6, and IL-10 (and sometimes IL-13), which induce B-cell
proliferation and differentiation into antibody-secreting plasma cells. IL-10
produced by T H2 cells inhibits
cytokine production by TH1
cells via the down-regulation of MHC expression by APCs. Conversely, IFN-γ produced by TH1 cells inhibits the
proliferation of TH2 cells (Figure 55–3).
Although these subsets have been well described in vitro, the nature of the
antigenic challenge that elicits
a
TH1 or TH2 phenotype
is less clear. Extracellular bacteria typi-cally cause the elaboration of TH2
cytokines, culminating in the production of neutralizing or opsonic antibodies.
In contrast, intracellular organisms (eg, mycobacteria) elicit the production
of TH1 cytokines, which lead to the activation of effector
cells such as macrophages. A less well-defined T-cell subset (TH3)
has been described that produces transforming growth factor-β (TGF-β), whose
numerous functions include down-regulation of prolifera-tion and
differentiation of T lymphocytes.
Recently,
a subset of CD4 T cells that secrete IL-17 (TH17)
has been implicated in neutrophil recruitment to sites of inflamma-tion. A
population of CD4 T cells that is essential for preventing autoimmunity and
allergy as well as maintaining homeostasis and tolerance to self antigens is
the regulatory (Treg) T cell. In the mouse, this cell population exists as
natural Treg (nTreg), derived directly from the thymus, and induced (adaptive)
Treg (iTreg), generated from naïve CD4 T cells in the periphery. Both
popula-tions have also been shown to inhibit antitumor immune responses and are
implicated in fostering tumor growth and progression. Recent attempts to
distinguish both populations have resulted in the discovery of a transcription
factor, Helios, in nTreg but not in iTreg cells.
CD8
T lymphocytes recognize endogenously processed pep-tides presented by
virus-infected cells or tumor cells. These pep-tides are usually
nine-amino-acid fragments derived from virus or protein tumor antigens in the
cytoplasm and are loaded onto MHC class I molecules (Figure 55–2) in the
endoplasmic reticu-lum. In contrast, class II MHC molecules present peptides
(usu-ally 11–22 amino acids) derived from extracellular (exogenous) pathogens
to CD4 T helper cells. In some instances, exogenous antigens, upon ingestion by
APCs, can be presented on class I MHC molecules to CD8 T cells. This
phenomenon, referred to as “cross-presentation,” involves retro-translocation
of antigens from the endosome to the cytosol for peptide generation in the
prote-osome and is thought to be useful in generating effective immune
responses against infected host cells that are incapable of priming T
lymphocytes. Upon activation, CD8 T cells induce target cell death via lytic
granule enzymes (“granzymes”), perforin, and the Fas-Fas ligand (Fas-FasL)
apoptosis pathways.
B lymphocytes undergo selection in the bone marrow, during which self-reactive B lymphocytes are clonally deleted while B-cell clones specific for foreign antigens are retained and expanded. The repertoire of antigen specificities by T cells is genetically determined and arises from T-cell receptor gene rear-rangement while the specificities of B cells arise from immuno-globulin gene rearrangement; for both types of cells, thesedeterminations occur prior to encounters with antigen. Upon an encounter with antigen, a mature B cell binds the antigen, inter-nalizes and processes it, and presents its peptide—bound to class MHC—to CD4 helper cells, which in turn secrete IL-4 and IL-5.
These interleukins
stimulate B-cell proliferation and dif-ferentiation into memory B cells and
antibody-secreting plasma cells. The primary antibody response consists mostly
of IgM-class immunoglobulins. Subsequent antigenic stimulation results in a
vigorous “booster” response accompanied by class (isotype) switching to produce
IgG, IgA, and IgE antibodies with diverse effector functions (Figure 55–3).
These antibodies also undergo affinity maturation, which allows them to bind
more efficiently to the antigen. With the passage of time, this results in
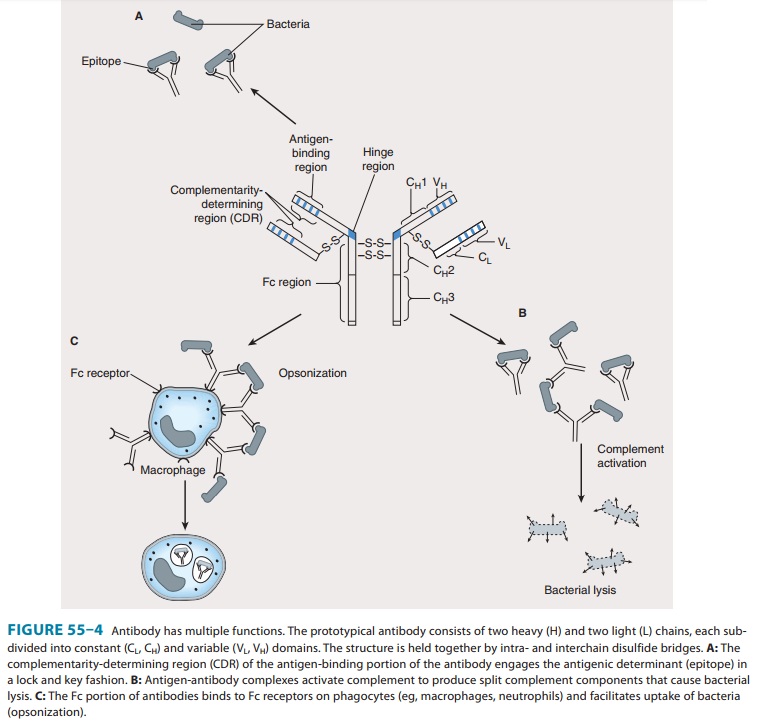
acceler-ated elimination of microorganisms in subsequent infections. Antibodies
mediate their functions by acting as opsonins to enhance phagocytosis and
cellular cytotoxicity and by activating complement to elicit an inflammatory
response and induce bacte-rial lysis (Figure 55–4).
Related Topics