Chapter: Basic & Clinical Pharmacology : Immunopharmacology
Abnormal Immune Responses
ABNORMAL IMMUNE RESPONSES
Whereas
the normally functioning immune response can success-fully neutralize toxins,
inactivate viruses, destroy transformed cells, and eliminate pathogens,
inappropriate responses can lead to extensive tissue damage (hypersensitivity)
or reactivity against self antigens (autoimmunity); conversely, impaired
reactivity to appro-priate targets (immunodeficiency) may occur and abrogate
essen-tial defense mechanisms.
Hypersensitivity
Hypersensitivity can
be classified as antibody-mediated or cell-mediated. Three types of
hypersensitivity are antibody-mediated (types I–III), while the fourth is
cell-mediated (type IV). Hypersensitivity occurs in two phases: the
sensitization phase and the effector phase. Sensitization occurs upon initial encounter
with an antigen; the effector phase involves immunologic memory and results in
tissue pathology upon a subsequent encounter with that antigen.
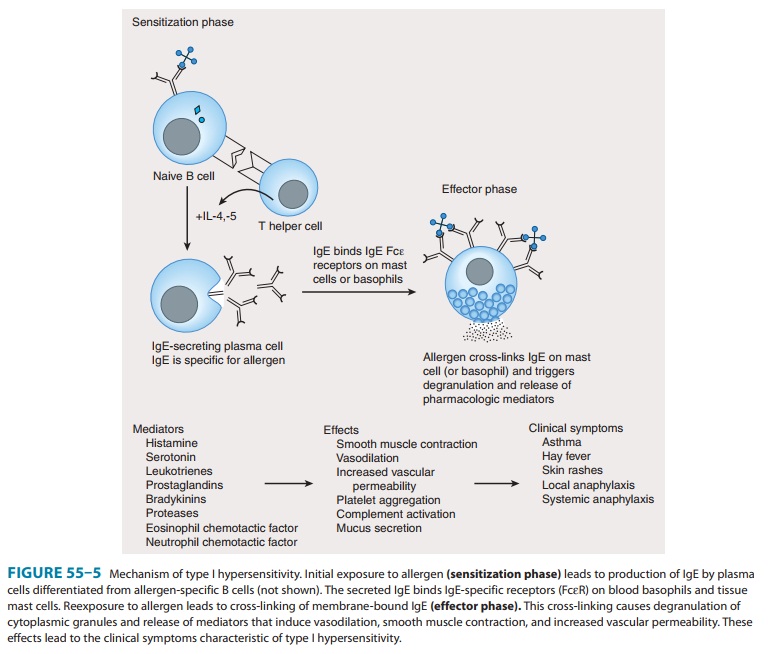
Type I—Immediate,
or type I, hypersensitivity is IgE-mediated,with symptoms usually occurring
within minutes following the patient’s reencounter with antigen. Type I
hypersensitivity results from cross-linking of membrane-bound IgE on blood
basophils or tissue mast cells by antigen. This cross-linking causes cells to
degran-ulate, releasing substances such as histamine, leukotrienes, and
eosinophil chemotactic factor, which induce anaphylaxis, asthma, hay fever, or
urticaria (hives) in affected individuals (Figure 55–5). A severe type I
hypersensitivity reaction such as systemic anaphylaxis (eg, from insect envenomation,
ingestion of certain foods, or drug hypersensitivity) requires immediate
medical intervention.
Type II—Type II hypersensitivity results from the formationof antigen-antibody complexes between foreign antigen and IgM or IgG immunoglobulins. One example of this type of hypersen-sitivity is a blood transfusion reaction that can occur if blood is not cross-matched properly. Preformed antibodies bind to red blood cell membrane antigens that activate the complement cas-cade, generating a membrane attack complex that lyses the trans-fused red blood cells. In hemolytic disease of the newborn, anti-Rh IgG antibodies produced by an Rh-negative mother cross the placenta, bind to red blood cells of an Rh-positive fetus, and damage them. The disease is prevented in subsequent pregnancies by the administration of anti-Rh antibodies to the mother 24–48 hours after delivery (see Immunosuppressive Antibodies, below).
Type II
hypersensitivity can also be drug-induced and may occur during the
administration of penicillin (for example) to allergic patients. In these
patients, penicillin binds to red blood cells or other host tissue to form a
neoantigen that evokes production of antibodies capable of inducing complement-mediated
red cell lysis. In some circumstances, subsequent administration of the drug
can lead to systemic anaphylaxis (type I hypersensitivity).
Type III—Type
III hypersensitivity is due to the presence ofelevated levels of
antigen-antibody complexes in the circulation that ultimately deposit on
basement membranes in tissues and vessels. Immune complex deposition activates
complement to produce components with anaphylatoxic and chemotactic activi-ties
(C5a, C3a, C4a) that increase vascular permeability andrecruit neutrophils to
the site of complex deposition. Complex deposition and the action of lytic
enzymes released by neutrophils can cause skin rashes, glomerulonephritis, and
arthritis in these individuals. If patients have type III hypersensitivity against
a particular antigen, clinical symptoms usually occur 3–4 days after exposure
to the antigen.
Type IV:
Delayed-type hypersensitivity— Unlike type I,II, and III hypersensitivities,
delayed-type hypersensitivity (DTH) is cell-mediated, and responses occur 2–3
days after exposure to the sensitizing antigen. DTH is caused by
antigen-specific DTH TH1
cells and induces a local inflammatory response that causes tissue damage
characterized by the influx of antigen-nonspecific
inflammatory cells, especially macrophages. These cells are
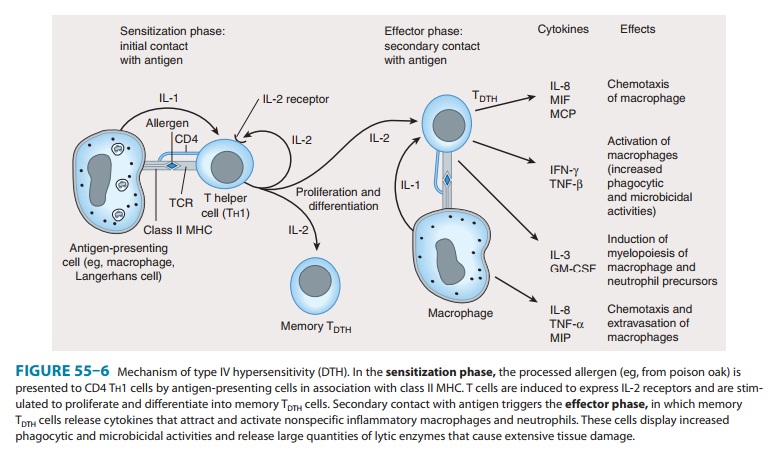
recruited
under the influence of TH1-produced cytokines (Figure 55–6), which chemoattract
circulating monocytes and neutrophils, induce myelopoiesis, and activate
macrophages. The activated macrophages are primarily responsible for the tissue damage associated
with DTH. Although widely considered to be deleterious, DTH responses are very
effective in eliminating infec-tions caused by intracellular pathogens such as
Mycobacterium tuberculosis and Leishmania species. Clinical manifestations of
DTH include tuberculin and contact hypersensitivities. Tuberculosis exposure is
determined using a DTH skin test. Positive responses show erythema and
induration caused by accu-mulation of macrophages and DTH T (TDTH) cells at the
site of the tuberculin injection. Poison ivy is the most common cause of
contact hypersensitivity, in which pentadecacatechol, the lipo-philic chemical
in poison ivy, modifies cellular tissue and results in a DTH T-cell response.
Autoimmunity
Autoimmune disease
arises when the body mounts an immune response against itself due to failure to
distinguish self tissues and cells from foreign (nonself ) antigens or loss of
tolerance to self. This phenomenon derives from the activation of self-reactive
T and B lymphocytes that generate cell-mediated or humoral immune responses
directed against self antigens. The pathologic conse-quences of this reactivity
constitute several types of autoimmune diseases. Autoimmune diseases are highly
complex due to MHC genetics, environmental conditions, infectious entities, and
dys-functional immune regulation. Examples of such diseases include rheumatoid
arthritis, systemic lupus erythematosus, multiple scle-rosis, and
insulin-dependent diabetes mellitus (type 1 diabetes). In rheumatoid arthritis,
IgM antibodies (rheumatoid factors) are pro-duced that react with the Fc
portion of IgG and may form immune complexes that activate the complement
cascade, causing chronic
In systemic lupus erythe-matosus, antibodies are made
against DNA, histones, red blood cells, platelets, and other cellular
components. In multiple sclerosis and type 1 diabetes, cell-mediated autoimmune
attack destroys myelin surrounding nerve cells and insulin-producing islet beta
cells of the pancreas, respectively. In type 1 diabetes, activated CD4 TDTH cells that infiltrate
the islets of Langerhans and recognize self islet beta cell peptides are
thought to produce cytokines that stimu-late macrophages to produce lytic
enzymes, which destroy islet beta cells. Autoantibodies directed against the
islet beta cell antigens are produced, but do not contribute significantly to
disease.
A number of mechanisms
have been proposed to explain auto-immunity:
Exposure of antigens previously sequestered
from the immune system (eg, lens protein, myelin basic protein) to
self-reactive T lymphocytes.
Molecular mimicry by invading pathogens, in
which immune responses are directed at antigenic determinants on pathogens that
share identical or similar epitopes with normal host tissue. This phenomenon
occurs in rheumatic fever following Streptococcus
pyogenes infection, in which heart damage isthought to arise from an immune
response directed against streptococcal antigens shared with heart muscle. The
suggested viral etiology of autoimmune diseases has been ascribed to immune
responses (both cell-mediated and humoral) directed against virus epitopes that
mimic self antigens.
Inappropriate expression of class II MHC molecules on the membranes of cells that normally do not express class II MHC (eg, islet beta cells). Increased expression of MHC II may increase presentation of self peptides to T helper cells, which in turn induce CTL, TDTH, and B-lymphocyte cells that react against self antigens.
Immunodeficiency Diseases
Immunodeficiency
diseases result from inadequate function in the immune system; the consequences
include increased susceptibility to infections and prolonged duration and
severity of disease. Immunodeficiency diseases are either congenital or arise
from extrinsic factors such as bacterial or viral infections or drug
treat-ment. Affected individuals frequently succumb to infections caused by
opportunistic organisms of low pathogenicity for the immuno-competent host.
Examples of congenitally acquired immunodefi-ciency diseases include X-linked
agammaglobulinemia, DiGeorge’s syndrome, and severe combined immunodeficiency
disease (SCID) due to adenosine deaminase (ADA) deficiency.
X-linked
agammaglobulinemia is a disease affecting males that is characterized by a
failure of immature B lymphocytes to mature into antibody-producing plasma
cells. These individuals are susceptible to recurrent bacterial infections,
although the cell-mediated responses directed against viruses and fungi are
preserved. DiGeorge’s syndrome is due to failure of the thymus to develop,
resulting in diminished T-cell responses (TDTH,
CTL), while the humoral response remains functional, but does not benefit from
T-cell help.
The ADA enzyme
normally prevents the accumulation of toxic deoxy-ATP in cells. Deoxy-ATP is
particularly toxic to lym-phocytes, and leads to death of T and B cells.
Absence of the enzyme therefore results in SCID. Infusion of the purified
enzyme ( pegademase, from bovine
sources) and transfer of ADA gene-modified lymphocytes have both been used
successfully to treat this disease.
AIDS
represents the classic example of immunodeficiency dis-ease caused by extrinsic
viral infection, in this instance the human immunodeficiency virus (HIV). This
virus exhibits a strong tro-pism for CD4 T helper cells; these become depleted,
giving rise to increased frequency of opportunistic infections and malignancies
in infected individuals. AIDS is also characterized by an imbalance in TH1
and TH2 cells, and the ratios of cells and their functions
are skewed toward TH2. This results in loss of
cytotoxic T-lymphocyte activity, loss of delayed hypersensitivity, and
hyper-gammaglobulinemia.
Related Topics