Chapter: Biotechnology Applying the Genetic Revolution: Inherited Defects
Mitochondrial Defects
MITOCHONDRIAL DEFECTS
The mitochondrial genome is a
tiny fraction of an animal cell’s genetic information. In terms of coding DNA
there are about 100 mb in the nuclear genome and only 15.4 kb in human
mitochondrial DNA (mtDNA). Consequently, the vast majority of mutations
affecting coding DNA occur in nuclear DNA. Furthermore, there are thousands of
copies of mitochondrial DNA in most cells, so a mutation in one gene on one
copy has little effect on the whole organism. Because each original mutation
must occur on a single molecule of mtDNA, the chances of its spreading in the
mitochondrial population and becoming fixed seem highly unlikely. Despite these
factors, the proportion of hereditary defects affecting mtDNA is surprisingly
large.
One contributing factor is
that mitochondrial DNA has a much higher mutation rate than nuclear DNA.
Oxidation damage to DNA, due to reactive oxygen species generated by the
respiratory chain, is much higher in mitochondria. In addition, there are fewer
repair systems in mitochondria and the mtDNA is not protected by histones.
Moreover, mitochondria go through many more divisions than the cells that
contain them. (Although animal mtDNA is evolving very fast, the situation in
plants is quite different.
Plant mtDNA is much larger
[150 kbp to 2.5 Mbp], contains introns, and evolves relatively slowly.)
Sperm cells contribute
nuclear DNA during fertilization but do not donate their mitochondria to the
zygote. Consequently, all the mitochondria in a particular individual are
derived from ancestors in the female egg cell or oocyte, which has approximately 100,000 copies of mtDNA. Despite
this, the rate at which mutations in mtDNA become established in the cell is
some 10-fold greater than for nuclear DNA. Apparently, there is a bottleneck
during the replication of mtDNA. When primordial germ cells develop into
oocytes, only a few of their mitochondria divide and give rise to progeny (Fig.
16.7). This allows mutations to spread through the mitochondrial population in
a specific cell line.
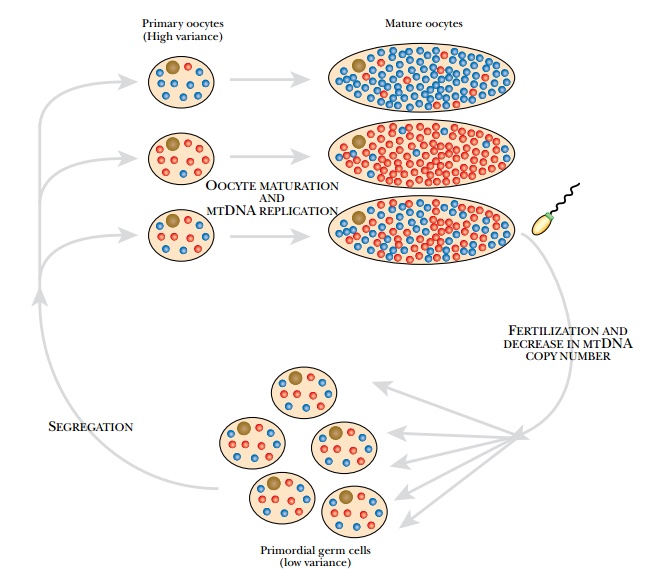

Disorders due to
mitochondrial mutations are maternally inherited. In addition, the population
of mitochondria within an individual may all carry the mutation, called homoplasmy, or there may be a mixture
of mutant and normal mitochondria, called
heteroplasmy. The severity of the defect will depend to some extent on the
proportion of defective
mitochondria, and this may vary among individuals with the same mutation.
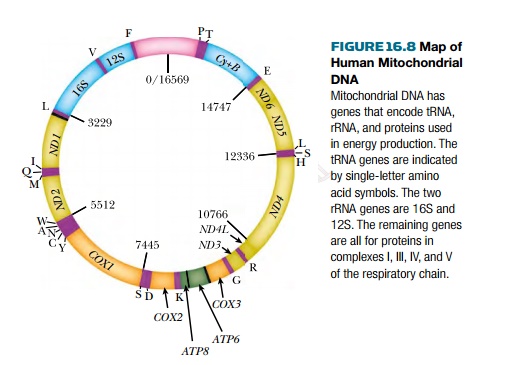
Mitochondria are highly specialized in generating energy via respiration, and the majority of mitochondrial genes affect the respiratory chain (Fig. 16.8). Consequently, almost all genetic defects in mtDNA affect respiration and result in lowered energy production. Because different tissues of the body differ greatly in energy requirements, mitochondrial defects affect different tissues to very different extents. Brain and muscle are especially susceptible because of their high energy consumption.
Neuropathy, ataxia, and
retinitis pigmentosa (NARP) syndrome is due to base changes at position 8993 of
the mtDNA molecule. These affect the ATP6
gene, encoding a component of the ATP
synthetase. This enzyme normally couples energy from the respiratory chain
to the synthesis of ATP. The result is a lack of ATP to energize the cell.
Nerve and muscle cells are most affected, and muscular weakness, dementia,
seizures, and sensory malfunction are among the results. Symptoms occur when
70% to 90% of the mitochondria are affected.
If more than 90% are
affected, death in infancy is the usual outcome.
Myoclonus epilepsy and ragged-red fibers (MERRF) syndrome is due to a defect in the mitochondrial lysine tRNA gene. Most cases are due to the point mutation A8344G (A to G at position 8344 of the mtDNA molecule). Defective tRNA results in decreased synthesis of several mitochondrial proteins and hence lowered respiration. Transient seizures, loss of muscle coordination, and loss of muscle cells result. The term ragged-red fibers refers to clumps of defective mitochondria that aggregate in muscle cells and were originally visualized by staining with a red dye. Symptoms are seen if 90% or more of the mitochondria in nerve and muscle cells are affected.
Related Topics