Chapter: Biotechnology Applying the Genetic Revolution: Inherited Defects
Genetic Screening and Counseling
GENETIC SCREENING AND COUNSELING
As explained earlier, close
relatives are more likely to produce malformed children because of harmful
recessive alleles coming together. It is also true that two people from
otherwise unrelated families that both have a history of the same hereditary
disease might be wise to avoid having children. Today we can do more than give
general advice. Testing of prospective parents for high-risk genetic diseases
can now be done for an increasing number of genes. The typical approach is to
use a hybridization probe or PCR analysis. This will reveal whether a mutant
copy of the gene is present in a DNA sample from the person tested. If marital
partners both test positive for a recessive defect, they will have to decide
whether or not to take the risk of having children. For a recessive defect,
where both parents are carriers, one in four children will get the disease.
The basic problem with
genetic screening is that our ability to detect genetic defects has far outrun
our ability to cure them. Thus, screening may reveal a defect about which
nothing can be done, and this may cause severe psychological distress to the
individuals concerned. For example, Huntington’s disease is an autosomal
dominant condition that results in movement disorder, dementia, and ultimately
death. The symptoms usually appear only in the 30s, although they may be
delayed as late as the 60s in some patients. Genetic screening helps those who
are afflicted decide whether or not to have children. It also provides them
with the knowledge that they will eventually develop an incurable and fatal
disease. Some cancers and coronary heart disease have a genetic component
(often multigenic). Screening may allow affected individuals to avoid factors
that tend to trigger the disease. On the other hand, especially if the
treatment is complicated and/or only partially effective, such knowledge may be
a burden.
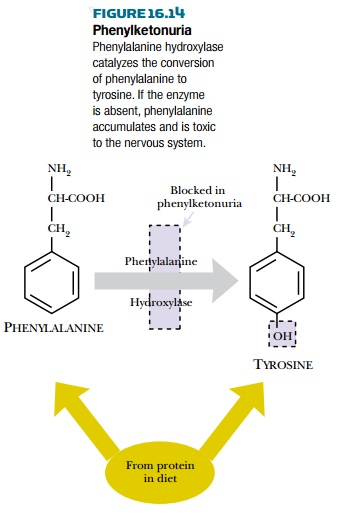
Genetic screening sometimes allows successful intervention by modifying diet or lifestyle. The classic case is phenylketonuria. The absence of the enzyme phenylalanine hydroxylase causes a buildup of phenylalanine and a deficiency of the product, tyrosine (Fig. 16.14). Excess phenylalanine is neurotoxic; if untreated, this condition results in severe damage to the central nervous system, and patients require permanent care for life. However, a diet low in phenylalanine avoids the buildup of this amino acid and largely alleviates the problems. Phenylketonuria occurs in 1 in 10,000 live births and is routinely screened for in developed nations using blood samples taken a week or so after birth. The diet may be relaxed somewhat later in life after development of the nervous system is largely complete.
Given that a reliable test is
available, it is generally agreed that neonatal screening is justified if the
disease is reasonably frequent (say 1 in 10,000 or more), the disease is
severely damaging, and that treatment is available that significantly improves
the condition.
It is also possible to
examine embryos during early pregnancy by drawing samples of amniotic fluid
which contain some cells from the fetus, a procedure called amniocentesis, or by examining
extraembryonic fetal tissue. These
cells may be genetically screened to see, for example, if the fetus is
homozygous for a recessive defect. Embryos doomed to grow up with hereditary
defects could then be aborted early if the parents wish to do so. It is also
now possible to screen embryos obtained by in
vitro fertilization before they are implanted. This avoids aborting of an
unwanted fetus should a serious genetic defect be discovered.
Related Topics