Chapter: Biotechnology Applying the Genetic Revolution: Inherited Defects
Cystic Fibrosis
CYSTIC FIBROSIS
About one in 2000 white
children suffer from cystic fibrosis.
This condition is due to homozygous recessive mutations. In other words two
defective copies of the gene, one from each parent, must be inherited for the
child to suffer from the disease. Humans with a single defective allele are
carriers but do not show symptoms, as a single wild-type version of the gene is
sufficient for normal health.
The protein encoded by the
cystic fibrosis gene is referred to as the CFTR
protein (for cystic fibrosis transporter) and is found in the cell membrane
where it acts as a channel for chloride ions. In healthy people, this channel
can be opened or shut as needed by the cell. The CFTR protein consists of a
series of membrane-spanning segments with a central control module (Fig. 16.9).
If the control module has a phosphate group attached, the channel is open, and
when the phosphate group is removed, it shuts.
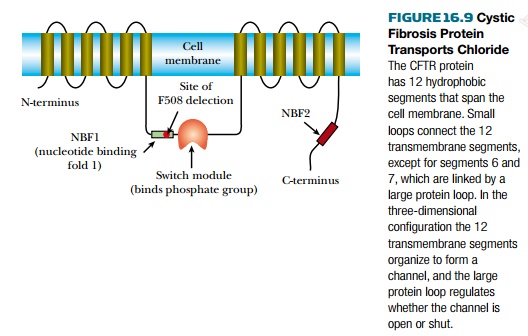
In cystic fibrosis patients,
control of the chloride channel is defective. This, in turn, affects a variety
of other processes. The most harmful is that the mucus that lines and protects
the lungs is abnormally thick. Lack of chloride ions leads to lack of
sufficient water accompanying the mucus. This not only causes obstructions but
also allows the growth of harmful microorganisms. Cells lining the airways of
the lungs are killed and replaced with fibrous scar tissue—hence the name of
the disease. Eventually the patient succumbs to respiratory failure.
The general location of the cystic fibrosis gene (to within 2 million base pairs) was found by screening large numbers of genetic markers (RFLPs) in members of families with a cystic fibrosis patient. The gene was found on the longer (q) arm of chromosome 7 between bands q21 and q31. After subcloning this region of the chromosome in large segments, the cystic fibrosis gene was found to occupy 250,000 base pairs and have 24 exons, encoding a protein of 1480 amino acids (Fig. 16.10). Because 1480 amino acids need only 4440 base pairs to encode them, this means that scarcely 2% of the cystic fibrosis gene is actually coding DNA. The rest consists of introns.
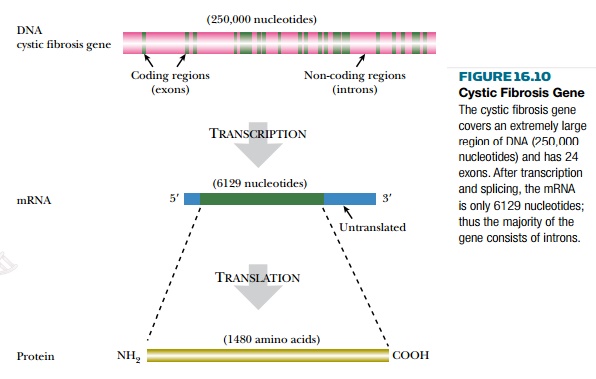
About 70% of the cystic fibrosis victims in North America share the same genetic defect. They all have a small deletion of three bases that code for the amino acid phenylalanine at position 508 of the normal protein. In Denmark, 90% of cystic fibrosis cases are due to this F508 deletion (250,000 nucleotides) (F = phenylalanine in single-letter code), whereas in the Middle East it accounts for only 30%. The other cystic fibrosis cases are the result of more than 500 different mutations, which makes genetic screening difficult.
The distribution and
relatively high frequency of the F508
deletion suggest that it originated in Western Europe, perhaps around 2000
years ago, and has been positively selected. Although two defective copies of
the CFTR gene are highly detrimental, heterozygotes with one F508 allele and one normal allele show only
moderate reduction of ion flow and water movement. This does not cause cystic
fibrosis but is sufficient to significantly reduce the loss of fluids during
diarrhea. Such heterozygotes are more resistant to the effects of enteric
infections such as typhoid and cholera that cause dehydration via extensive
diarrhea.
Related Topics