Chapter: Modern Pharmacology with Clinical Applications: Drug Absorption and Distribution
Mechanisms of Solute Transport Across Membranes
MECHANISMS OF
SOLUTE TRANSPORT ACROSS MEMBRANES
Except for intravenous
administration, all routes of drug administration require that the drug be
trans-ported from the site of administration into the systemic circulation. A
drug is said to be absorbed only when it has entered the blood or lymph
capillaries.
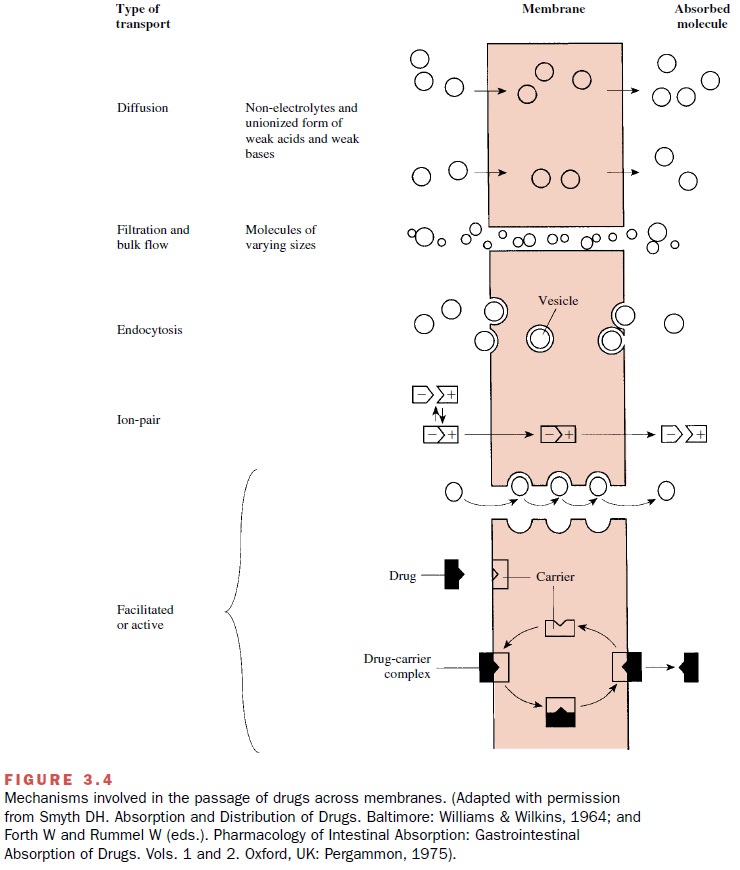
The trans-port of drugs across membranes entails one or more of the following processes: (1)
passive diffusion, (2) filtra-tion, (3) bulk flow, (4) active transport, (5)
facilitated transport, (6) ion pair transport, (7) endocytosis, and exocytosis
(Fig. 3.4). These processes also participate in the transport of substances
necessary for cellular maintenance and growth.
Passive Diffusion
Most drugs pass through
membranes by passive diffu-sion (down their concentration gradient) of the un-ionized moiety. The rate of diffusion
depends mainly on the lipid–water
partition coefficient rather than on lipid solubility per se. For example, the
central nervous sys- tem depressant barbital is almost completely un-ionized at
physiological pH and therefore should be able to cross membranes easily.
However, barbital’s lipid–water partition coefficient is sufficiently low that
diffusion across membranes proceeds at an extremely slow rate. This slow rate
of passage across central nervous system (CNS) membranes largely explains why
the time of on-set (latent period) of
drug action after barbital adminis-tration is delayed.
A drug will accumulate in the membrane until the ratio of its concentration in the membrane and its concentration in the extracellular fluid equal its parti-tion coefficient. A concentration gradient is thereby established between the membrane and the intracel-lular space; this gradient is the driving force for the passive transfer of the drug into the cell. Thus, a drug that has a very high lipid–water partition coefficient will have a large concentration gradient, and this fa-vors its rapid diffusion across the membrane and into the cell.
Filtration
The rate of filtration
depends both on the existence of a pressure gradient as a driving force and on
the size of the compound relative to the size of the pore through which it is
to be filtered. In biological systems, the pas-sage of many small water-soluble
solutes through aque-ous channels in the membrane is accomplished by
fil-tration. The hypothetical diameter of these pores is about 7 Å, a size that
generally limits passage to com-pounds of molecular weight less than 100 (e.g.,
urea, ethylene glycol).
Bulk Flow
Most substances, lipid
soluble or not, cross the capillary wall at rates that are extremely rapid in
comparison with their rates of passage across other body mem-branes. In fact,
the supply of most drugs to the various tissues is limited by blood flow rather
than by restraint imposed by the capillary wall. This bulk flow of liquid occurs through intercellular pores and is the
major mechanism of passage of drugs across most capillary en-dothelial
membranes, with the exception of those in the CNS.
Active Transport
The energy-dependent movement
of compounds across membranes, most often against their concentration
gra-dient, is referred to as active
transport. In general, drugs will not be actively transported unless they
sufficiently resemble the endogenous substances (such as sugars, amino acids,
nucleic acid precursors) that are the nor-mal substrates for the particular
carrier system. This transport involves the reversible binding of the mole-cule
to be transferred to a membrane component (a car-rier) of complementary
configuration.
Several mechanisms of active
transport have been postulated. One transport model proposes that the drug
molecule combines with a specific mobile carrier (Fig. 3.4), probably a
protein, on one side of the membrane. The complex formed diffuses across the
membrane to the opposite side, where the complex dissociates, thus releasing
the drug into the aqueous compartment bor-dering the opposite membrane surface.
The carrier pro-tein can then return to its initial side to bind more drug.
Another model involves a chainlike arrangement of sites in transport channels
to which the drug can bind. The drug would be transferred from one site to
another until it had traversed the membrane.
Active transport of a
particular substance occurs in one direction only. The number of molecules
trans-ported per unit of time will reach a maximum (Tm) once the
binding capacity of the carrier becomes saturated. Drugs such as levodopa (for
parkinsonism) and α-methyldopa (for hypertension) are actively transported.
Since active transport often
requires energy in the form of adenosine triphosphate (ATP), compounds or
conditions that inhibit energy production (e.g., iodoac-etate, fluoride, cyanide,
anaerobiosis) will impair active transport. The transport of a given compound
also can be inhibited competitively by the coadministration of other compounds
of sufficient structural similarity that they can compete with the first
substance for sites on the carrier protein.
Facilitated Diffusion
The transfer of drugs by
facilitated diffusion has many of the characteristics associated with active
transport, including being a protein carrier–mediated transport system that
shows saturability and selectivity. It differs from active transport, however,
in that no energy input is required beyond that necessary to maintain normal
cellular function. In facilitated transport the movement of the transported
molecule is from regions of higher to regions of lower concentrations, so the driving force for facilitated transport is the concentration gradient. Although
the initial rate of drug transfer will be pro-portional to the magnitude of the
concentration gradi-ent, at some point further increases in drug concentra-tion
no longer increase the transport rate; that is, Tm has been reached, since the binding sites on the
carrier are now completely saturated.
Ion Pair Transport
Absorption of some highly
ionized compounds (e.g., sulfonic acids and quaternary ammonium compounds) from
the gastrointestinal tract cannot be explained in terms of the transport
mechanisms discussed earlier. These compounds are known to penetrate the lipid
membrane despite their low lipid–water partition coef-ficients. It is
postulated that these highly lipophobic drugs combine reversibly with such
endogenous com-pounds as mucin in the gastrointestinal lumen, forming neutral
ion pair complexes; it is this neutral complex that penetrates the lipid
membrane by passive diffu-sion.
Endocytosis
Endocytosis involves the cellular uptake of exogenous molecules or complexes inside plasma membrane– derived vesicles.
This process can be divided into two major categories: (1) adsorptive or
phagocytic uptake of particles that have been bound to the membrane sur-face
and (2) fluid or pinocytotic uptake, in which the particle enters the cell as
part of the fluid phase. The solute within the vesicle is released
intracellularly, pos-sibly through lysosomal digestion of the vesicle mem-brane
or by intermembrane fusion (Fig. 3.4).
Related Topics