Chapter: Modern Pharmacology with Clinical Applications: Drug Absorption and Distribution
Absorption of Drugs From the Alimentary Tract
ABSORPTION OF
DRUGS FROM THE ALIMENTARY TRACT
Oral Cavity and Sublingual Absorption
In contrast to absorption
from the stomach and intes-tine, drugs absorbed from the oral cavity enter the
gen-eral circulation directly. Although the surface area of the oral cavity is
small, absorption can be rapid if the drug has a high lipid–water partition
coefficient and therefore can readily diffuse through lipid membranes. Since
the diffusion process is very rapid for un-ionized drugs, pKa will be a major determinant
of the lipid– water partition coefficient for a particular therapeutic agent.
For instance, the weak base nicotine (pKa
8.5) reaches peak blood levels four times faster when ab-sorbed from the mouth
(pH 6), where 40 to 50% of the drug is in the un-ionized form, than from the
gastroin-testinal tract (pH 1–5), where the drug exists mainly in its ionized
(protonated) form.
Although the oral mucosa is
highly vascularized and its epithelial lining is quite thin, drug absorption
from the oral cavity is limited. This is due in part to the rela-tively slow
dissolution rate of most solid dosage forms and in part to the difficulty in
keeping dissolved drug in contact with the oral mucosa for a sufficient length
of time. These difficulties may be overcome if the drug is placed under the
tongue (sublingual administration) or between the cheek and gum (buccal cavity)
in a formu-lation that allows rapid tablet dissolution in salivary se-cretions.
The extensive network of blood vessels facili-tates rapid drug absorption.
Sublingual administration is the route of choice for a drug like nitroglycerin
(glyc-eryl trinitrate), whose coronary vasodilator effects are required quickly
in cases of angina. Furthermore, if swallowed, the drug would be absorbed from
the gas-trointestinal tract and carried to the liver, where nitro-glycerin is
subject to rapid metabolism and inactivation.
Absorption from the Stomach
Although the primary function of the stomach is not ab-sorption, its rich
blood supply and the contact of its con-tents with the epithelial lining of the
gastric mucosa provide a potential site for drug absorption. However, since
stomach emptying time can be altered by many variables (e.g., volume of
ingested material, type and viscosity of the ingested meal, body position,
psycholog-ical state), the extent of gastric absorption will vary from patient
to patient as well as at different times within a single individual.
The low pH of the gastric
contents (pH 1–2) may have consequences for absorption because it can
dra-matically affect the degree of drug ionization. For ex-ample, the weak base
diazepam (pKa 3.3) will be
highly protonated in the gastric juice, and consequently, ab- sorption across
lipid membranes of the stomach will be particularly slow. On the other hand,
the weak acid acetaminophen (pKa
9.5) will exist mainly in its un-ionized form and can more readily diffuse from
the stomach into the systemic circulation (Fig. 3.3).
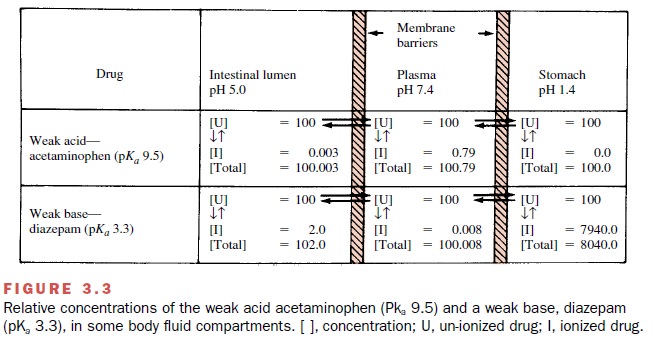
Because of the influence of
pH on ionization of weak bases, basic drugs may be trapped in the stomach even
if they are administered intravenously. Since basic compounds exist primarily
in their un-ionized form in the blood (pH 7.4), they readily diffuse from the
blood into the gastric juice. Once in contact with the gastric contents (pH
1–2), they will ionize rapidly, which re-stricts their diffusibility. At
equilibrium, the concentra-tion of the un-ionized lipid-soluble fraction will
be iden-tical on both sides of the gastric membranes, but there will be more total basic drug on the side where
ioniza-tion is greatest (i.e., in gastric contents). This means of drug
accumulation is called ion trapping.
Absorption from the Small Intestine
The epithelial lining of the
small intestine is composed of a single layer of cells called enterocytes. It
consists of many villi and microvilli and has a complex supply of blood and
lymphatic vessels into which digested food and drugs are absorbed. The small
intestine, with its large surface area and high blood perfusion rate, has a
greater capacity for absorption than does the stomach. Most drug absorption occurs in the proximal jejunum (first 1–2 m in
humans).
Although transfer of drugs
across the intestinal wall can occur by facilitated transport, active
transport, en-docytosis, and filtration, the predominant process for most drugs
is diffusion. Thus, the pKa
of the drug and the pH of the intestinal fluid (pH 5) will strongly influ-ence
the rate of drug absorption. While weak acids like phenobarbital (pKa 7.4) can be absorbed from
the stom-ach, they are more readily absorbed from the small in-testine because
of the latter’s extensive surface area.
Conditions that shorten
intestinal transit time (e.g., diarrhea) decrease intestinal drug absorption,
while in-creases in transit time will enhance intestinal absorption by
permitting drugs to remain in contact with the intes-tinal mucosa longer.
Although delays in gastric empty-ing time will increase gastric drug
absorption, in gen-eral, total drug
absorption may actually decrease, since material will not be transferred to the
large absorptive surface of the small intestine.
Absorption from the Large Intestine
The large intestine has a
considerably smaller absorp-tive surface area than the small intestine, but it
may still serve as a site of drug absorption, especially for com-pounds that
have not been completely absorbed from the small intestine. However, little
absorption occurs from this site, since the relatively solid nature of the
in-testinal contents impedes diffusion of the drug from the contents to the mucosa.
The most distal portion of
the large intestine, the rectum, can be used directly as a site of drug
adminis-tration. This route is especially useful where the drug may cause
gastric irritation, after gastrointestinal sur-gery, during protracted
vomiting, and in uncooperative patients (e.g., children) or unconscious ones.
Dosage forms include solutions and suppositories. The processes involved in rectal
absorption are similar to those de-scribed for other sites.
Although the surface area
available for absorption is not large, absorption can still occur, owing to the
ex-tensive vascularity of the rectal mucosa. Drugs ab-sorbed from the rectum
largely escape the biotransfor-mation to which orally administered drugs are
subject, because a portion of the blood
that perfuses the rectum is not
delivered directly to the liver, and therefore, rectally administered drug, at
least in part, escapes hepatic first-pass metabolism.
Related Topics