Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Sporozoa
Malaria
MALARIA
Malaria is a febrile illness caused by a parasitic infection of human erythrocytes transmitted by the bite of a mosquito. The fevers are accompanied by headache, sweats, malaise, and typically appear in paroxysmal episodes lasting hours and re- curring for weeks. Complications due to capillary blockade can be fatal, particu- larly in the brain.
EPIDEMIOLOGY
Malaria has a worldwide distribution between 45°N and 40°S latitude, generally at alti-tudes below 1800μm. P. vivax is the most widely distributed of the four species, and to-gether with the uncommon P. malariae, is found primarily in temperate and subtropical areas. P. falciparum is the dominant organism of the tropics. P. ovale is rare and found principally in Africa.
The intensity of malarial transmission in an endemic area depends on the density and feeding habits of suitable mosquito vectors and the prevalence of infected humans, who serve as parasite reservoirs. In hyperendemic areas (areas where more than half of the population is parasitemic), transmission is usually constant, and disease manifestations are moderated by the development of immunity. Mortality is largely restricted to infants and to nonimmune adults who migrate into the region. When the prevalence of disease is lower, transmission is typically intermittent. In this situation, solid immunity does not develop and the population suffers repeated, often seasonal, epidemics, the impact of which is shared by people of all ages.
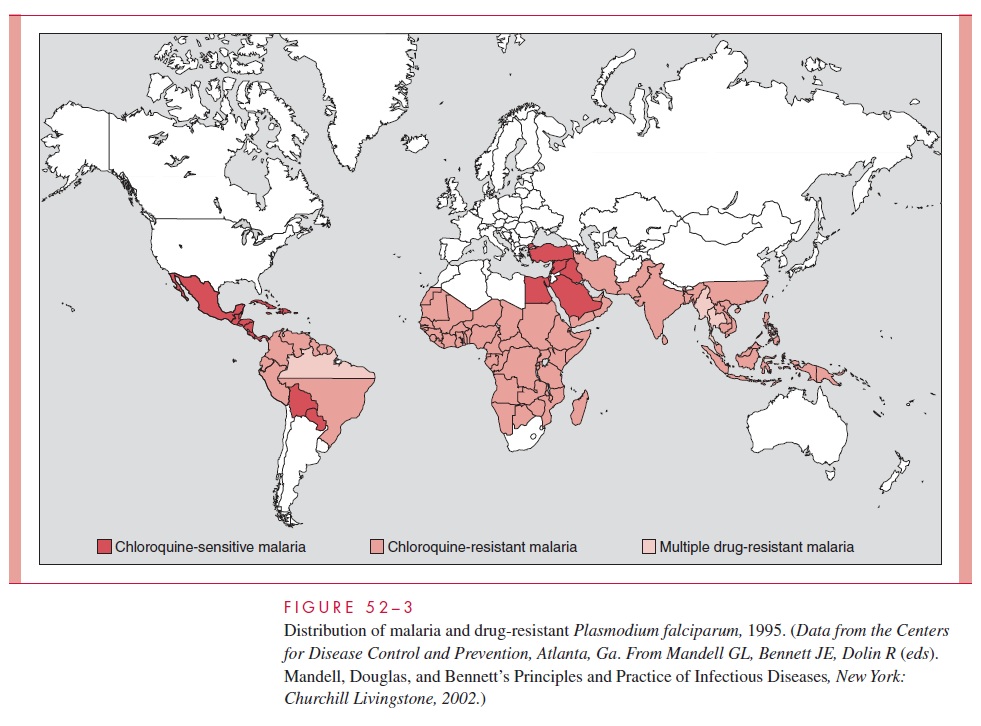
Presently, it is estimated that 2 billion people live in malaria endemic areas in 103 of the poorest countries of Africa, Asia, Latin America, and Oceania (Fig 52–3). Between 25 and 50% of these persons are thought to be carrying the malaria parasite at any given time. From 1 to 3 million individuals, primarily African children, die of this disease annually. A recent study concluded that the development of resistance to chloroquine, the single most widely used antimalarial agent, has increased mortality four- to eight-fold. Although endemic malaria disappeared from the United States three decades ago, imported cases continue to be reported, and the recent worldwide resurgence of malaria combined with an increase in international travel has resulted in an increase in the num-ber of US cases to approximately 1000 annually.
Forty-five percent of patients with im-ported malaria have acquired the disease in Africa, 30% in Asia, and 10% in the Caribbean or Latin America. Fifty percent of recent infections have involved American travelers: nearly 60% of these acquired their infection in Africa. Clinical manifestations typically develop within 6 months of arrival of cases in the United States; however, one fourth of cases caused by P. vivax are delayed beyond that time. Approximately 40% of imported cases and almost all associated fatalities have been caused by the virulent P.falciparum. Tragically, most of these cases could have been prevented or successfullytreated. Congenital malaria in infants born in the United States of mothers from malari-ous areas is occasionally observed. Infections transmitted by transfusions of whole blood, leukocytes, or platelets, or by organ transplantation are, fortunately, now unusual in this country due to the improved screening procedures of blood banks. Anopheline mosquitoes capable of transmitting malaria are present in the United States, and, rarely, malaria is transmitted from an imported case to individuals who have never traveled out-side of the country.
PATHOGENESIS
The fever, anemia, circulatory changes, and immunopathologic phenomena characteristic of malaria are all the result of erythrocytic invasion by the plasmodia.
Fever
Fever, the hallmark of malaria, appears to be initiated by the process of RBC rupture that leads to the liberation of a new generation of merozoites (sporulation). To date, all attempts to detect the factor(s) mediating the fever have been unsuccessful. It is possi-ble that parasite-derived pyrogens are released at the time of sporulation; alternatively, the fever might result from the release of interleukin-1 (IL-1) and/or tumor necrosis factor (TNF) from macrophages involved in the ingestion of parasitic or erythrocytic debris. Early in malaria, RBCs appear to be infected with malarial parasites at several different stages of development, each inducing sporulation at a different time. The resulting fever is irregular and hectic. Because temperatures in excess of 40°C destroy mature parasites, a single population eventually emerges, sporulation is synchronized, and fever occurs in distinct paroxysms at 48-hour or, in the case of P. malariae, 72-hour intervals. Periodicity is seldom seen in patients who are rapidly diagnosed and treated.
Anemia
Parasitized erythrocytes are phagocytosed by a stimulated reticuloendothelial system or are destroyed at the time of sporulation. At times, the anemia is disproportionate to the degree of parasitism. Depression of marrow function, sequestration of erythrocytes within the enlarging spleen, and accelerated clearance of nonparasitized cells all appear to con-tribute to the anemia. The mechanisms responsible for the latter are unclear. Intravascular hemolysis, although uncommon, may occur, particularly in falciparum malaria. When he-molysis is massive, hemoglobinuria develops, resulting in the production of dark urine. This process in conjunction with malaria is known asblackwater fever.
Circulatory Changes
The high fever results in significant vasodilatation. In falciparum malaria, vasodilatation leads to a decrease in the effective circulating blood volume and hypotension, which may be aggravated by other changes in the small vessels and capillaries. The intense para-sitemias P. falciparum is capable of producing and the adhesion of infected RBCs to the endothelium of visceral capillaries can impair the microcirculation and precipitate tissue hypoxia, lactic acidosis, and hypoglycemia. Although all deep tissues are involved, the brain is the most intensely affected.
Cytokines
Elevated levels of IL-1 and TNF are consistently found in patients with malaria. Probably released at the time of sporulation, these proteins are certainly an essential part of the host’s immune response to malaria . By modulating the effects of endothelial cells, macrophages, monocytes, and neutrophils, they may play an important role in the destruction of the invading parasite. However, TNF levels increase with parasite density and high concentrations appear harmful. TNF has been shown to cause upregulation of endothelial adhesion molecules; high concentrations might precipitate cerebral malaria by increasing the sequestration of P. falciparum–parasitized erythrocytes in the cerebral vas-cular endothelium. Alternatively, excessive TNF levels might precipitate cerebral malaria by directly inducing hypoglycemia and lactic acidosis.
Other Pathogenic Phenomena
Thrombocytopenia is common in malaria and appears to be related to both splenic pooling and a shortened platelet lifespan. Both direct parasitic invasion and immune mechanisms may be responsible. There may be an acute transient glomerulonephritis in falciparum malaria and progressive renal disease in chronic P. malariae malaria. These phenomena probably result from the host immune response, with deposition of immune complexes in the glomeruli.
IMMUNITY
Once infected, the host quickly mounts a species- and strain-specific immunologic response that typically limits parasite multiplication and moderates the clinical manifestations of dis-ease, without eliminating the infection—a phenomenon referred to as premunition. A pro-longed recovery period marked by recurrent exacerbations in both symptoms and number of erythrocytic parasites follows. With time, these recrudescences become less severe and less frequent, eventually stopping altogether.
The exact mechanisms involved in this recovery are uncertain. In simian and probably in human malaria, recovery is known to require the presence of both T and B lympho-cytes. It is probable that the T lymphocytes act partially through their helper effect on an-tibody production. Some authorities have suggested that they also play a direct role through lymphokine production by stimulating effector cells to release nonspecific factors capable of inhibiting intraerythrocytic multiplication. The B lymphocytes begin produc-tion of stage- and strain-specific antiplasmodial antibodies within the first 2 weeks of par-asitemia. With the achievement of high levels of antibodies, the number of circulating parasites decreases. The infrequency with which malaria occurs in young infants has been attributed to the transplacental passage of such antibodies. It is uncertain whether they are directly lethal, act as opsonizing agents, or block merozoite invasion of RBCs.
In simian malaria, the parasite can undergo antigenic variation and thereby escape the suppressive effect of the antibodies. This antigenic variation leads to cycles of recrudes-cent parasitemia but ultimately to production of specific antibodies to the variants, and cure. It seems probable that similar changes occur in humans, leading to the eventual dis-appearance of erythrocytic parasites. With P. falciparum and P. malariae, which have no persistent hepatic forms, this results in cure. With P. falciparum,the disease typically does not exceed 1 year, but with P. malariae the erythrocytic infection can be extremely persistent, lasting in one case up to 53 years. How erythrocytic parasites circulating in numbers too small to be detected on routine blood films escape immunologic destruction remains a puzzle. In a closely related simian malaria, splenectomy results in rapid cure, suggesting that suppressor T lymphocytes in the spleen may play a protective role. In in-fection with P. vivax and P. ovale, latent hepatic infection may result in the discharge of fresh merozoites into the bloodstream after the disappearance of erythrocytic forms. This phenomenon, known as relapse, is capable of maintaining infection for 3 to 5 years.
Related Topics