Chapter: Obstetrics and Gynecology: Common Medical Problems in Pregnancy
Hematologic Disease
HEMATOLOGIC DISEASE
Anemia
The plasma and cellular
composition of blood change sig-nificantly during pregnancy, with the expansion
of plasma volume proportionally greater than that of the red blood cell mass.
On average, there is a 1000-mL increase in plasma volume and a 300-mL increase
in red-cell volume (a 3:1 ratio). Because the hematocrit (Hct) reflects the
pro-portion of blood made up primarily of red blood cells, Hct demonstrates a
“physiologic” decrease during pregnancy; therefore, this decrease is not
actually an anemia.
Anemia in
pregnancy is generally defined as an Hct less than 30% or a hemoglobin of less
than 10 g/dL.
The direct fetal consequences of
anemia are minimal, although infants born to mothers with iron deficiency may
have diminished iron stores as neonates. The maternal consequences of anemia
are those associated with any adult anemia. If anemia is corrected, the woman
with an ade-quate red-cell mass enters labor and delivery better able to
respond to acute peripartum blood loss and to avoid the risks of blood or blood
product transfusion.
IRON-DEFICIENCY ANEMIA
Iron-deficiency
anemia is by far the most frequent type ofanemia seen in
pregnancy, accounting for more than 90% of cases. Because the iron content of
the standard American diet and the endogenous iron stores of many American
women are not sufficient to provide for the increased iron requirements of
pregnancy, the National Academy of Sciences recommends 27 mg of iron
supplementation (present in most prenatal vitamins) daily for pregnant women.
Most prescription prenatal vitamin/mineral prepa-rations contain 60 to 65 mg of
elemental iron.
All pregnant women should be
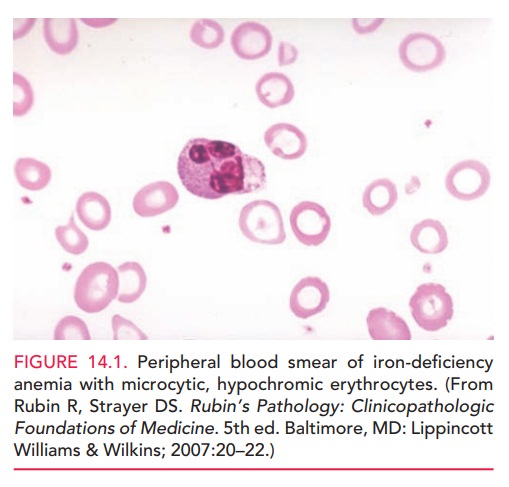
screened for iron-deficiency anemia. Severe iron-deficiency anemia is
charac-terized by small, pale erythrocytes (Fig. 14.1) and red-cell indices
that indicate a low mean corpuscular volume and low mean corpuscular hemoglobin
concentration. Addi-tional laboratory studies usually demonstrate decreased
serum iron levels, an increased total iron-binding capacity, and a decrease in
serum ferritin levels. A recent dietary history is obviously important,
especially if pica (the con-sumption of non-nutrient substances such as starch,
ice, or dirt) exists. Such dietary compulsions may contribute to iron
deficiency by decreasing the amount of nutritious food and iron consumed.
Treatment of iron-deficiency
anemia generally requires an additional 60 to 120 mg of elemental iron per day,
in ad-dition to the iron in the prenatal vitamin/mineral prepa-ration. Iron
absorption is facilitated by or with vitamin C supplementation or ingestion
between meals or at bed-time on an empty stomach. The response to therapy is
first seen as an increase in the reticulocyte count approximately 1 week after
starting iron therapy. Because of the plasma expansion associated with
pregnancy, the Hct may not increase significantly, but rather stabilizes or
increases only slightly.
FOLATE DEFICIENCY
Adequate intake of folic acid (folate) has been found to reduce the risk of neural tube defects (NTDs) in the fetus
The first occurrence of NTDs may
be reduced by as much as 36% if women of reproductive age consume 0.4 mg of
folate daily both before conception and during the first trimester of
pregnancy. The Recommended Daily Dietary Allowance for folate for pregnant
women is 0.6 mg. Folate deficiency is especially likely in multiple gestations
or when patients are taking anticonvulsive medications. Women with a history of
a prior NTD-affected pregnancy or who are being treated with anticonvulsive
drugs may reduce the risk of NTDs by more than 80% with daily intake of 4 mg of
folate in the months in which conception is attempted and for the first
trimester of pregnancy.
Folate is found in green leafy
vegetables and is now an added supplement in cereal, bread, and grain
prod-ucts. These supplements are designed to enable women to easily consume 0.4
mg to 1 mg of folate daily. Prescription prenatal vitamin/mineral preparations
contain 1 mg of folic acid.
OTHER ANEMIAS
The hemoglobinopathies are a heterogenous group of single-gene
disorders that includes the structural hemoglo-bin variants and the
thalassemias. Hereditary
hemolyticanemias are also rare causes of anemia in pregnancy.Some examples
are hereditary spherocytosis, an autosomal dominant defect of the erythrocyte
membrane; glucose 6-phosphate dehydrogenase deficiency; and pyruvate kinase
deficiency.
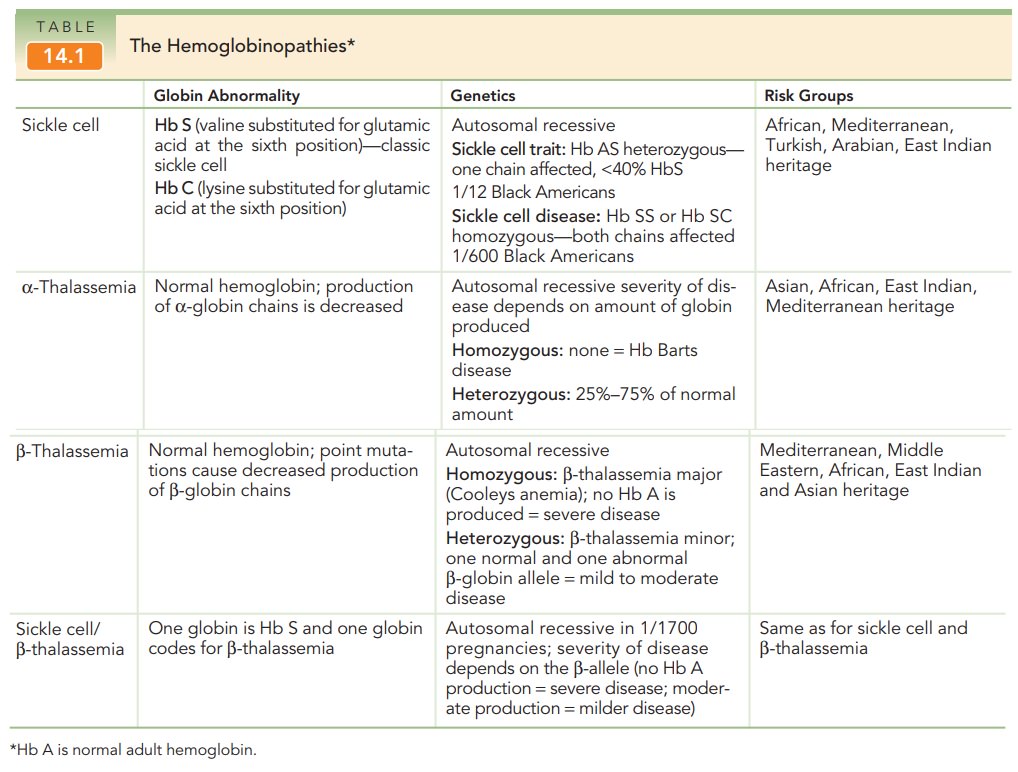
The Hemoglobinopathies
More than 270 million people
worldwide are heterozygous carriers of hereditary disorders of hemoglobin, and
at least 300,000 affected homozygotes or compound homozygotes are born each
year. The hemoglobinopathies include the thalassemias (α-thalassemia, β-thalassemia) and the sickle cell
spectrum: sickle cell trait (Hb AS), sickle cell disease (Hb SS), and sickle
cell disorders (Hb SC and sickle cell β-thalassemia) (Table 14.1).
Hemoglobin (Hb) consists of four
interlocking poly-peptide chains, each of which has an attached heme mol-ecule.
The polypeptide chains are called alpha, beta, gamma, delta, epsilon, and zeta.
Adult hemoglobins con-sists of two alpha chains and either two beta chains (Hb
A), two gamma chains (Hb F), or two delta chains (Hb A2). The beta
chains are the oxygen-carrying subunits of the hemoglobin molecule. Hb F is the
primary hemoglobin of the fetus from 12 to 24 weeks of gestation. In the third
trimester, production of Hb F decreases as production of beta-chains and Hb A
begins.
α-thalassemia is generally caused by missing copiesof the α-globin gene; however, occasionally point muta-tions can cause functional abnormalities in the protein. Humans normally have 4 copies of the α-globin gene. Those with 3 copies are asymptomatic, those with 2 copies have mild anemia, and those with 1 copy have hemolytic anemia. Individuals in whom the gene is absent have Hb Barts disease, which results in hydrops fetalis and intra-uterine death.
Phenotypic expressions of -thalassemia vary because of the many
possible mutations in the β-globin
gene. Some mutations cause an absence of the protein, while others result in a
defective globin protein. β-thalassemia
major occurs in homozygotes and is a severe disease, whereas diagnosis of β-thalassemia minor
(heterozygotes) may include asymptomatic to clinically anemic patients.
The sickle cell disorders are autosomal recessive dis-orders caused by
point mutations that lead to functional abnormalities in the β-globin chains. Instead of normal
Hb A, individuals with this disorder have abnormal Hb S. Hb S is unstable,
especially under conditions of low oxy-gen tension. The unstable Hb S causes a
structural change resulting in deformity of the normal spheroid shape of the
red blood cell into the shape of a “sickle.” These abnor-mally shaped cells
lead to increased viscosity, hemolysis, and a further decrease in oxygenation.
Sickling that occurs in small blood vessels can cause a vaso-occlusive crisis, in which the blood supply to vital organs is
compromised.
Heterozygotic individuals (Hb AS)
have sickle celltrait and are
asymptomatic. The most severe form of thedisease, which occurs in homozygotic
individuals (Hb SS), is called sickle cell
anemia. Sickle cell disorders are found not only in patients who have Hb
SS, but also in those who have Hb S and one other abnormality of β-globin struc-ture. The most
common are Hb SC disease and Hb S/β-thalassemia.
Women of
Mediterranean, Southeast Asian, or African descent are at higher risk of being
carriers for hemoglobinopathies and should be offered carrier screening.
If both parents are deemed to be
carriers of any hemoglo-binopathy, genetic counseling is recommended. For indi-viduals of non African descent,
initial testing should be done by complete blood count (CBC). Because
individuals of African de-scent are at high risk for carrying a gene for sickle
cell disease, these women should be offered hemoglobin electrophoresis in
addition to a CBC. Solubility testing, such as tests for thepresence of Hb
S (Sickledex), isoelectronic focusing, and high-performance liquid
chromatography (HPLC) are in-adequate for screening and fail to identify
important trans-missible hemoglobin gene abnormalities affecting fetal outcome.
Although the course of pregnancy
can vary according to the type of hemoglobinopathy, there is also individual
variation among patients with the same type of disorder. Besides the genetic
implications, patients with the sickle cell trait (Hb AS) have an increased
risk of urinary infec-tions but experience no other pregnancy complications.
Pregnancies in patients with HbS/β–thalassemia
are gen-erally unaffected. Patients who are Hb SS or Hb SC, in contrast, may
suffer vaso-occlusive episodes. Infections are also more common due to
functional asplenia caused by repetitive end-organ damage to the spleen.
Infection should be ruled out before attributing any pain to a vaso-occlusive
crisis.
Although prophylactic maternal
red cell transfusions for women with hemoglobinopathies have been used in the
past, transfusions are, for the most
part, reserved for com-plications of hemoglobinopathies such as congestive
heart failure, sickle cell disease crises unresponsive to hydration and
analgesics, and severely low levels of hemoglobin. Because fetal
outcomessuch as preterm labor, intrauterine growth restriction, and low birth
weight are more common in women with hemo-globinopathies, except those with
sickle cell trait, antenatal assessment of fetal well-being and growth is an
important part of managing patients with hemoglobinopathies.
Related Topics