Chapter: Medical Immunology: AIDS and Other Acquired Immunodeficiency Diseases
HIV and the Immune System
HIV and the Immune System
The primary cellular targets of HIV are CD4+ cells, particularly the helper T cells. Most infected CD4+ lymphocytes co-express the CD45RO marker, considered as activated or memory helper T lymphocytes. It thus appears that activated CD4+ T cells are most sus-ceptible to infection, but productive infection of resting CD4+ T cells has also been demon-strated. The infection of these cells involves several steps:
· The initial interaction of HIV and CD4+ cells involves specific regions of gp120 and the CD4 molecule. It is believed that that interaction results in a conformational change of gp120, which allows gp120 to interact with a co-receptor.
· Two β-chemokine receptors play the role of principal co-receptors. One, CCR-5 is the receptor for three chemokines: RANTES and macrophage inhibitory proteins 1α (MIP-1α ) and 1β (MIP-1β ). CCR-5 is expressed primarily by macrophages. The second, CXCR-4 (fusin), is expressed primarily by T lymphocytes.
· The interaction between CD120 and the co-receptor molecules results in exposure of the fusogenic domain of gp41, which can then interact with the cell membrane. This, in turn, results in the fusion of the membrane and the viral envelope and penetration of the nucleocapsid into the cytoplasm.
Different HIV strains seem to show different co-receptor specificities. Macrophage-tropic HIV strains (which predominate in the early stages of infection) use CCR-5 as co-re-ceptors. CD4 lymphotropic strains use CXCR-4 as co-receptors. Some strains that can in-fect both CD4 T lymphocytes and macrophages use CCR5 to infect macrophages, and a third chemokine receptor, CCR2b, to infect lymphocytes. Mucosal transmission involves the infection of both submucosal CD4+ T cells and Langerhans cells, followed by rapid spread to the regional lymph nodes, where it propagates to other CD4+ cells, including helper T cells, macrophages, and dendritic cells. When the virus is directly introduced into the blood stream, it will most likely be filtered in the spleen, where its ultimate fate will de-pend on its affinity for the different co-receptors mentioned above.
During HIV infection, the expression of co-receptors may be upregulated in cell pop-ulations that normally do not express them and that may allow HIV to infect those cell pop-ulations. However, some cell populations infected by HIV (e.g., intestinal cells) are not known to express CD4. The infection of mucosal epithelial cells is believed to be acquired from infected T cells and probably involves penetration through alternative receptors.
Once HIV enters a cell, its RNA is reverse transcribed and the resulting DNA inte-grates into the host genome, where its expression seems to depend on cellular activation. Several mechanisms of T-cell activation leading to enhanced replication of integrated HIV have been proposed:
· Infected macrophages and CD4+ lymphocytes are activated by concurrent infections (venereal or not).
· Some viral components may act as superantigens, interacting directly with the Vβ re-gions of specific types of T-cell receptors, consequently activating those cells.
· Dendritic cells in the submucosa and lymphoid tissues appear to bind HIV to their surface without becoming infected. Thus, the activation of HIV-carrying den-dritic cells could lead to clustering with noninfected T cells, which would re-ceive activating signals and HIV virus from the dendritic cells.
· Tumor necrosis factor (TNF) and interleukin-6 (IL-6) have the capacity to activate HIV replication in monocytes and T lymphocytes. In T lymphocytes, TNF in-duces the synthesis of a DNA-binding protein that binds to a nuclear factor kB (NFkB) site on the HIV-LTR, activating the expression of the integrated genome. A significant correlation between the levels of circulating viral RNA and of TNF and its soluble receptors supports the intimate relationships be-tween TNF release and HIV genome expression. As for the source of the TNF, there is evidence suggesting that HIV-infected macrophages release increased amounts of TNF and IL-6.
As HIV replicates, a vigorous anti-HIV immune response is elicited. A strong hu-moral immune response against HIV can be detected in most patients. Neutralizing anti-bodies, which inhibit the infectivity of free HIV in vitro, directed against epitopes of gp120 and gp41, can be demonstrated. Also potentially protective are ADCC-promoting antibod-ies, which react with gp160 expressed on the membrane of infected cells. The rate of pro-gression to AIDS and the mortality rate are considerably higher in individuals lacking neu-tralizing antibodies. However, neutralizing antibodies do not prevent infected individuals from eventually developing AIDS, in part due to the high frequency of mutations in gp120, which result in the developing of mutants not neutralized by previously existing antibod-ies. It has also been reported that the virus in vivo does not expose the immunodominant regions of gp120, thus reducing the ability of antibodies to react with the viral particles. It has also been suggested that patients may develop enhancing antibodies. These antibodies react with gp41 and promote HIV infectivity by an unknown mechanism. In some studies, the presence of HIV-enhancing antibodies appears to be correlated with progression to-wards AIDS. Thus, the general consensus is that the humoral response elicited by HIV does not eliminate the infection and does not prevent evolution to AIDS.
Cell-mediated immune responses involve MHC–I–restricted CD8+ T lymphocytes, which recognize a variety of epitopes in gag, env, nef, and pol HIV proteins, expressed in association with MHC-I proteins. Cell-mediated cytotoxic reactions seem to be especially prominent in HIV-positive individuals who remain asymptomatic for prolonged periods of time. CD8 lymphocytes are also able, at least in vitro, to release cytokines (particularly RANTES, MIP-α and β, and a recently discovered interleukin, IL-16), which appear to act by blocking the chemokine co-receptors used by HIV to penetrate noninfected CD4+ cells. In addition, a soluble factor released by CD8+ , CD28+ T cells inhibits the replication of in-tegrated HIV. Thus, cell-mediated immunity seems able to either block infection or reduce viral replication to levels tolerated by the immune system.
Most HIV-infected patients develop AIDS at a time in which there is still evidence of a strong antiviral antibody response. Several factors may contribute to the “escape” of HIV from the immune response mounted by the patient:
· HIV mutates at a much faster rate than most other viruses. This is due to the fact that the reverse transcriptase is error-prone and lacks copy-editing capabilities. The mutations affecting the epitopes of gp120 against which neutralizing antibod-ies are directed represent a selective advantage to the mutant, able to avoid recognition by the preformed antibodies.
· The virulence of HIV may change during the course of the infection (e.g., emergence of syncytia-inducing strains).
· HIV causes downregulation of HLA-A and HLA-B expression without affecting the expression of HLA-C and HLA-E. In this way the infected cells may escape de-tection by cytotoxic T lymphocytes but are not affected by NK cells.
· Humoral immune responses are relatively inefficient in eliminating viral-infected cells. ADCC and lysis of viral-infected cells after exposure to antibody and complement have been observed in vitro, but it is questionable that these may be significant defense mechanisms in vivo.
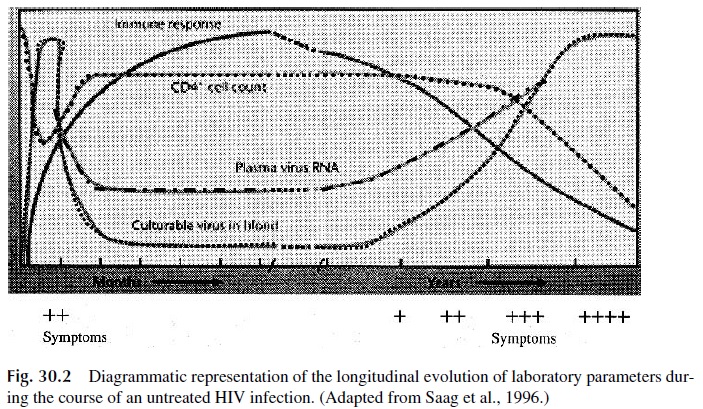
As the infection persists and progresses, there is a steady decline in the numbers of CD4+ cells (Fig. 30.2). This T-cell depletion affects predominantly the CD4, CD45RO+ population and is the direct cause of the profound immunodepression seen in AIDS pa-tients. HIV has been shown to be able to infect T-lymphocyte precursors. A major conse-quence of the infection of these targets is a significant decrease in the ability to repopulate the peripheral CD4 T-lymphocyte pool under attack by HIV. The result of excessive loss and lack of production of CD4 cells is a marked decrease in their absolute numbers.
Several factors have been suggested to account for the depletion of CD4+ T cells.
1. Direct cytotoxicity caused by virus replication is believed to be the most impor-tant cause of T-cell death. Accumulation of unintegrated DNA in the cytoplasm of infected cells is associated with vigorous HIV replication and cell death.
2. The cross-linking of CD4 molecules by gp120 is believed to prime T lympho-cytes for apoptosis. In this case, active infection may not be essential, since apop-tosis-primed T cells can undergo apoptosis when activated by some other stim-ulus, at least in vitro.
3. The expression of gp120 with unique sequences of the V1 to V2 and of the V3 re-gions of gp120 on the membrane of infected T cells facilitates the formation of syncytia by interaction and fusion with the membranes of noninfected cells ex-pressing CD4. The formation of syncytia allows direct cell-cell transmission of the virus and contributes to the reduction in the number of viable T cells. The emergence of strains with the syncytia-inducing sequences in infected patients is usually a late event in the course of an HIV infection, and associated with a faster progression to AIDS (median of 23 months), in part because these HIV strains are more apt to infect CD4 T-cell precursors.
4. The immune response against viral-infected T cells (mediated both by cytotoxic T cells and by ADCC mechanisms) may also contribute to CD4+ T-cell depletion.
5. Co-infection of HIV-infected T cells with other microorganisms, such as cy-tomegalovirus or Mycoplasma fermentans, has synergistic effects in the induc-tion of viral replication and cell death.
Several other factors beyond the depletion of CD4+ cells seem to contribute to the state of marked immunodepression associated with full-blown AIDS:
1. A reduction of the TcRβ variable region repertoire on T cells, reducing the num-ber of antigens to which the patient can mount an adequate immune response.
2. An imbalance of the TH1 and TH 2 subsets seems to precede the evolution to-wards AIDS. Several factors may contribute to this dysfunction, particularly lackof expression of CD80/86 by HIV-infected APCs. The lack of co-stimulatory signals involving CD80/86 and CD28 blocks TH1 differentiation and leads to predominant TH 2 activity. TH 2 cells release IL-4 and IL-10, which further down-regulate TH1 cells as well as the release of suppressor cytokines by CD8+ T lym-phocytes. It must be noted that clear evidence for predominant TH 2 activity in patients evolving towards symptomatic HIV infection has not been substantiated by a variety of investigators.
3. Release of soluble gp120 by infected cells. Soluble gp120 binds to CD4 and may block the interaction of this molecule with MHC-II antigens, therefore prevent-ing the proper stimulation of helper T cells by antigen-presenting cells.
4. Immune complexes involving viral antigens and the corresponding antibodies may also play a role in depressing immune responses. For example, binding of gp120:anti-gp120 complexes to CD4+ molecules of normal lymphocytes results in blocking T-cell activation via the TcR.
5. Infected monocytes are functionally abnormal, unable to perform chemotaxis, synthesize cytokines, and present antigens to helper T cells.
6. Of note is the fact that humoral immune responses become severely dysfunc-tional in patients with AIDS. These patients are unable to respond to antigenic challenges, while at the same time producing a variety of autoantibodies (in-cluding antinuclear autoantibodies and autoantibodies directed against platelets and lymphocytes). The synthesis of autoantibodies is probably a consequence of a state of permanent polyclonal activation of the B-cell system, probably as a re-sult of increased release of IL-6 by activated APC and T cells. At the same time, the de novo induction of protective immune responses is compromised by the lack of adequate T-cell help.
In contrast with the cytotoxic effect of HIV infection on activated T cells, the infec-tion of resting memory T cells, monocytes, macrophages, and related cells is not cytotoxic, and the infected cells become HIV reservoirs. Monocytes, macrophages, and related cells undergo chronic productive infection and perpetuate the infection in lymphoid tissues (lymph nodes and peri-intestinal lymphoid tissue). In contrast, HIV does not replicate on resting memory T cells. These cells can harbor the virus for long periods of time, are not affected by antiretroviral therapy, but are able to support HIV replication when activated.
Related Topics