Chapter: Genetics and Molecular Biology: Biological Assembly, Ribosomes and Lambda Phage
Global Structure of Ribosomes
The Global Structure of Ribosomes
The topic of in
vivo assembly or in vitro
reassembly cannot be studied without some knowledge of the
structure being assembled. Learning the structure of ribosomes has been a
tantalizing problem. Most of the interesting details of ribosomes are too small
to be seen by electron microscopy, but ribosomes are too large for application
of the conven-tional physical techniques such as nuclear magnetic resonance or
other spectroscopic methods. Although crystals of ribosomes have been formed
for X-ray crystallography, it will be a very long time until these yield
detailed structural information.
One of
the first questions to resolve about ribosome structure is the folding of rRNA.
It could be locally folded, which means that all nucleotides near each other in
the primary sequence are near each other in the tertiary structure (Fig. 21.1),
or it could be nonlocally folded. Recall that many proteins, IgG for instance,
are locally folded and contain localized domains, each consisting of a group of
amino acids contiguous in the primary sequence. Ribosomal RNA possesses a
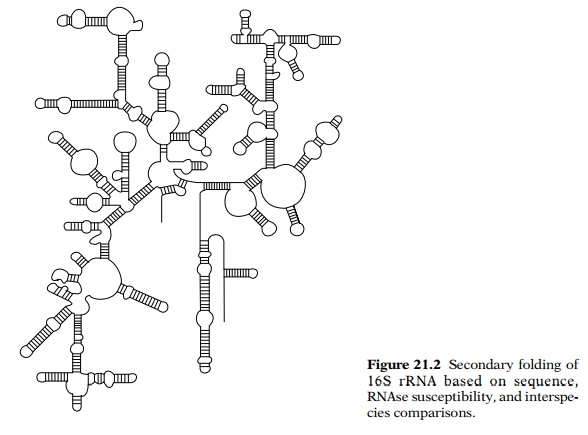
se-quence that permits extensive folding, most of which is local but a part of
which is nonlocal. Many stretches of the RNA can form double-helical hairpins.
The remainder is single-stranded in the form of small or medium sized regions
separating double-stranded regions of comple-mentary sequences (Fig. 21.2).
Several lines of evidence support the secondary structure which is predicted
primarily on the basis of se-quence. One of the most compelling is interspecies
or phylogenetic sequence comparisons. Frequently, a stretch A1,
which is postulated to base pair with stretch B1, is altered between
species. In such a case, the sequences of A2 and B2 in
the second species are both found to be altered
so as to
preserve the complementarity. Such compensating sequence changes are a strong
sign that the two stretches of nucleotides are base paired in both organisms
(Fig. 21.3). Additional evidence supporting the secondary structure predictions
is the locations of sites susceptible to cleavage by RNAses or to crosslinking.
The cleavage sites of RNAse A lie in loops or in unpaired regions. Crosslinking
portions of the rRNA with psoralen, a chemical which will intercalate into
helical DNA or RNA and covalently link to nucleotides in the presence of UV
light, joins regions postulated to be paired and also connects nucleotides that
are in close proximity because of the tertiary structure of the ribosome.
One of
the most obvious methods for obtaining structure information on ribosomes is to
look at them in the electron microscope. Due to its small size, a ribosomal
subunit looks like an indistinct blob. One can,
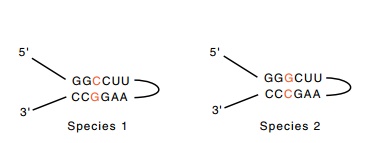
Figure
21.3 How base differencesin two regions
of RNA strengthen proposals for base pairing between the regions involved.
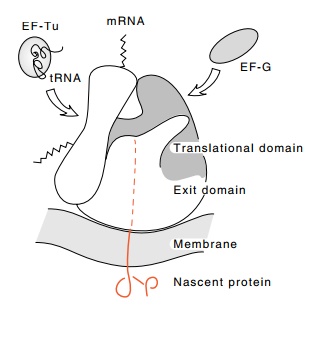
Figure
21.4 Diagrammatic repre-sentation of
the exit and translational domains of the ribo-some and their orientations with
respect to the membrane binding site. The nascent protein is shown as an
unfolded, extended chain during its passage through the ri-bosome. (Adapted
from Bernabeu and Lake, Proc. Nat. Acad. Sci. USA 79, 3111-3115 [1982].)
however, see several discernible shapes of blob,
suggesting that the subunit is not symmetrical and that it binds to the support
in only a few discrete orientations, much like a book resting on a table. As a
result, it is possible to average a number of images using electronic image
processing. It is even possible to subtract the averaged image of a ribosome
from the averaged image of a ribosome plus tRNA to reveal the binding location
of a tRNA molecule.
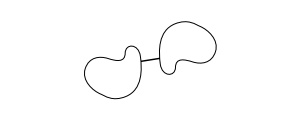
Individual ribosomal proteins can also be marked with antibodies and located by microscopy. IgG is bivalent, so it
will join two ribosomal subunits. Owing to the asymmetry of the subunits, the
position at which the two are linked by the antibody can often be ascertained.
Not only can many ribosomal proteins be located, but the exit site of nascent
polypeptide chains has been found through the use of antibodies to a
nonribosomal protein whose synthesis can be highly induced (Fig. 21.4). Other
features located by this method are the 5’ and 3’ ends of the rRNAs, dimethyl A
residues, translation factor-binding sites, and tRNAs.
Related Topics