Chapter: Biotechnology Applying the Genetic Revolution: Transgenic Plants and Plant Biotechnology
Functional Genomics in Plants
FUNCTIONAL
GENOMICS IN PLANTS
Because the complete DNA
sequences of rice, poplar tree, and Arabidopsis are known, plant scientists are
following functional genomics strategies. Rather than working on one specific
gene, the entire genome can be screened. Most of this type of work is still
done with Arabidopsis, but some has now moved into crop species such as rice,
corn, and soybeans. Functional genomics in plants uses a variety of techniques,
some of which have been discussed previously, but most rely on the removal or
blockage of gene expression. Novel genes and metabolic pathways useful for
understanding basic plant physiology are being analyzed in the hope of
improving our current crops.
Insertions are one method to
find the function of new genes. Transposons or T-DNA insertions are two methods
used to generate plant mutants. Here, instead of including a transgene, the
T-DNA or transposon includes only a reporter gene. When the T-DNA or transposon
integrates into the plant chromosome, the insertion may disrupt a plant gene.
When the insertion knocks out the function of a plant gene, the resulting
phenotype can be screened and assessed. By cloning the regions upstream and
downstream of the insertion, the plant gene that corresponds to the phenotype
can be identified.
Gene silencing is another
method to identify the function of plant genes. As described, gene silencing by
RNA interference (RNAi) is a phenomenon that was originally described in
plants. RNAi is triggered by double-stranded RNA, which is cut into short
segments (siRNA, short-interfering RNA). The RISC enzyme complex uses siRNA to
identify homologous RNA (in particular mRNA) and cut it up. This prevents mRNA
from being expressed into protein. This is exploited in the laboratory by
transforming a plant with small oligonucleotides that stimulate RISC to abolish
the expression of a chosen gene. The plant can then be assessed for any visible
phenotype associated with the gene knockdown.
Another method for generating
gene knockouts is fast neutron
mutagenesis. This uses fast neutrons
to induce DNA deletions. Fast neutrons are created by nuclear processes
such as nuclear fission, where free
neutrons with a kinetic energy close to 1 MeV are generated. These neutrons
cause deletions in exposed DNA. Therefore, seeds from the plant of interest
exposed to fast neutrons acquire random mutations. The dose of fast neutrons
and, consequently, the number of deletions per genome can be controlled. Seeds
treated with fast neutrons, known as M1 seeds, are grown into plants. Each
plant has a different deletion or set of deletions and a potentially different
phenotype.
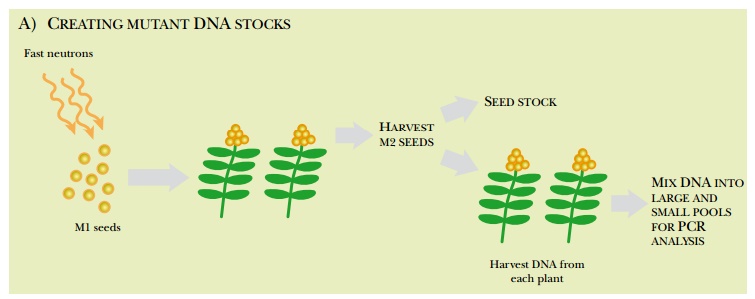
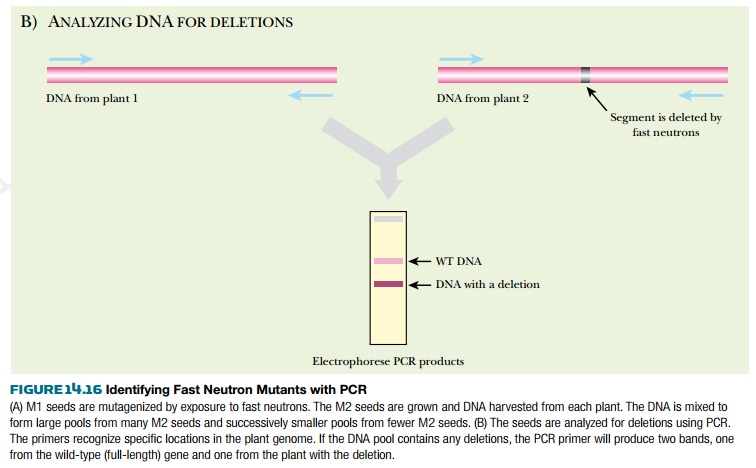
The seeds from each of these plants, called the M2 seeds, are collected. Most are saved as seed stock; the remaining M2 seeds are grown into plants. The DNA from the plants is isolated and collected into pools of varying sizes. For example, if the original pool had DNA from 100 plants, successively smaller pools of the 100 plants are made, down to just one or two plants per pool. These are usually screened by PCR to find specific genes with deletions. PCR primers are made to amplify a target gene from the largest pool of DNA. If a deletion was generated within the target gene in one of the plants, the PCR primers will amplify two bands, the wild-type gene plus a shorter segment from the deleted gene. The smaller DNA pools are then screened for the deletion until a specific M2 seed can be associated with the genetic deletion (Fig. 14.16).
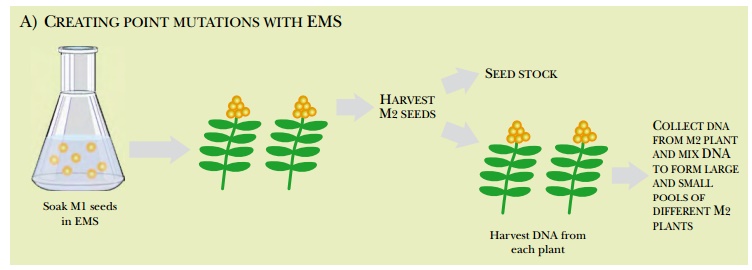
Yet another method of creating plant mutations is called TILLING (targeting-induced local lesions in genomes; Fig. 14.17). First, point mutations are created in a collection of seeds by soaking them in a chemical mutagen, such as EMS (ethyl methane sulfonate), which induces G/C and A/T transitions in DNA. As before, the M1 seeds are grown into plants and the second-generation seeds (M2) are mainly saved as stocks. Some M2 seeds are grown and the DNA is harvested and pooled into a large megapool and successively smaller pools as described earlier. PCR primers are used to amplify selected regions of the DNA. The PCR primers carry fluorescent labels. Consequently, the PCR products are labeled with two different labels, one at either end.
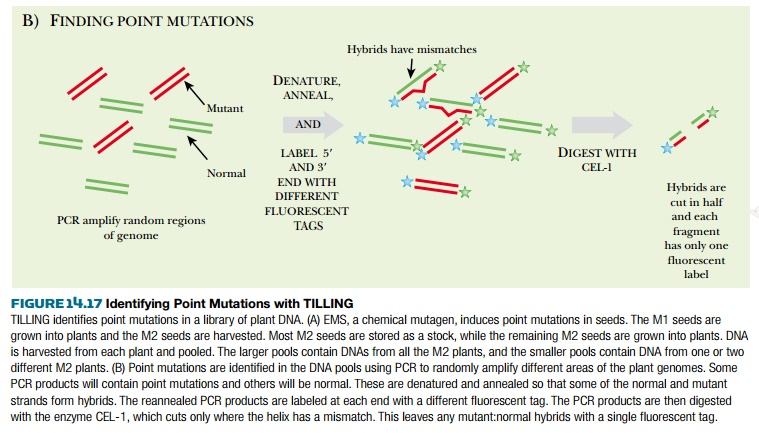
The key to identifying point mutations (as opposed to deletions) is to create heteroduplexes of mutant and wild-type DNA. Therefore, the PCR products are denatured to single strands and then slowly cooled so that the DNA strands reanneal. During reannealing, some mutant strands will anneal with wild-type strands and the heteroduplex will have a mismatched nucleotide. The enzyme CEL-1 cleaves mismatched DNA. If the PCR product is cleaved by CEL-1, it will have only one fluorescent label, whereas uncleaved DNA (with no mismatches) will have both fluorescent labels. When the PCR products are separated by gel electrophoresis, the digested mutant strands can be identified.
Related Topics