Chapter: Clinical Dermatology: Bullous diseases
Epidermolysis bullosa
Epidermolysis
bullosa
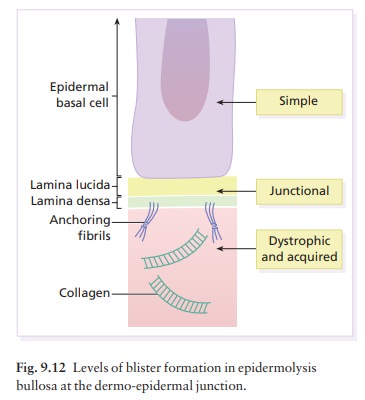
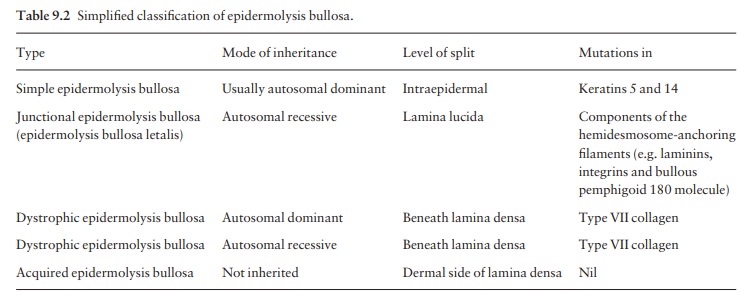
There are many types of epidermolysis bullosa: the five main ones are listed in Table 9.2. All are charac-terized by an inherited tendency to develop blisters after minimal trauma, although at different levels in the skin (Fig. 9.12). The more severe types have a catastrophic impact on the lives of sufferers. Acquired epidermolysis bullosa is not inherited and was discussed earlier.
Simple epidermolysis bullosa
Several subtypes are recognized, of which the most common are the Weber–Cockayne (mainly affecting the hands and feet) and the Dowling–Meara (featur-ing herpetiform blisters on the trunk) types. Most are inherited as autosomal dominant conditions and are caused by abnormalities in genes responsible for pro-duction of the paired keratins (K5 and K14) expressed in basal keratinocytes (see Fig. 2.4). Linkage studies show that the genetic defects responsible for the most common types of simple epidermolysis bullosa lie on chromosomes 17 and 12.
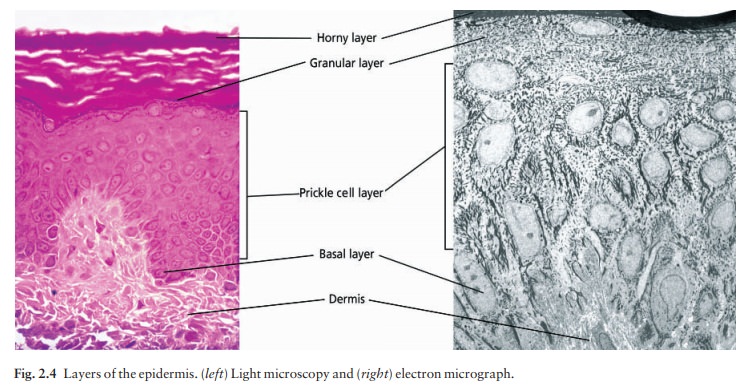
Blisters
form within or just above the basal cell layers of the epidermis and so tend to
heal without scarring. Nails and mucosae are not involved. The problems are
made worse by sweating and ill-fitting shoes. Blistering can be minimized by
avoiding trauma, wearing soft well-fitting shoes and using foot powder. Large
blisters should be pricked with a sterile needle and dressed. Their roofs
should not be removed. Local antibiotics may be needed.
Junctional epidermolysis bullosa
The
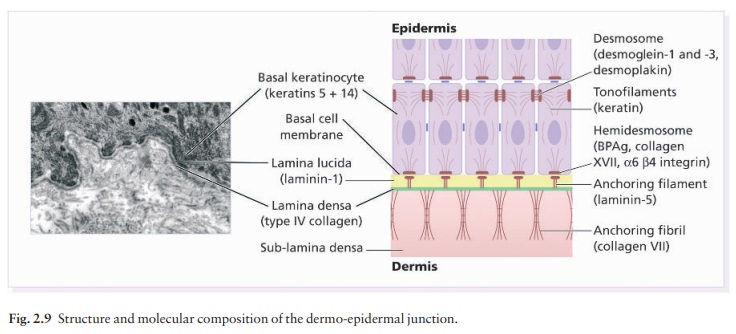
abnormalities in the basal lamina include loss of anchoring filaments and defective
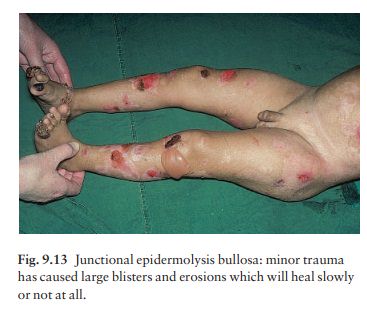
laminins (; see Fig. 2.9). This rare and often lethal condition is evident at
birth. The newborn child has large raw areas and flaccid blisters, which are
slow to heal (Fig. 9.13). The peri-oral and peri-anal skin is usually involved,
as are the nails and oral mucous membrane. There is no effective systemic
treatment. Hopes for the future include adding the normal gene to epidermal
stem cells, and then layering these onto the denuded skin.
Dystrophic epidermolysis bullosa
There are many subtypes, all of which probably result from abnormalities of collagen VII, the major struc-tural component of anchoring fibrils
Autosomal dominant dystrophic epidermolysis bullosa
In
this type blisters appear in late infancy. They are most common on friction
sites (e.g. the knees, elbows and fingers), healing with scarring and milia
formation. The nails may be deformed or even lost. The mouth is not affected.
The only treatment is to avoid trauma and to dress the blistered areas.
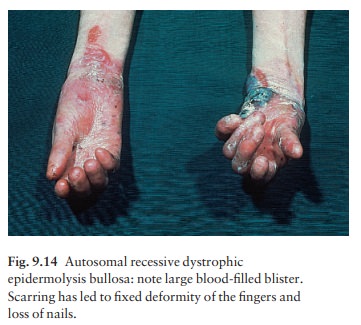
Autosomal recessive dystrophic epidermolysis bullosa
In
this tragic form of epidermolysis bullosa, blisters start in infancy. They are
subepidermal and may be filled with blood. They heal with scarring, which can
be so severe that the nails are lost and webs form between the digits (Fig.
9.14). The hands and feet may become useless balls, having lost all fingers and
toes. The teeth, mouth and upper part of the oesophagus are all affected;
oesophageal strictures may form. Squamous cell carcinomas of the skin are a
late complication. Treatment is unsatisfactory. Phenytoin, which reduces the
raised dermal collagenase levels found in this variant, and systemic steroids
are disappointing. It is especially important to minimize trauma, to prevent
contractures and web formation between the digits, and to combat anaemia and
secondary infection. Referral to centres with expertise in management of these
patients is strongly recommended.
Related Topics